GLIVEC
GLIVEC ® 400 mg
Imatinib.
IDENTIFICATION DU MEDICAMENT
FORMES ET PRÉSENTATIONS
Comprimé pelliculé jaune très foncé à brun orangé, ovaloïde, biconvexe, abords biseautés, portant l'inscription « NVR » sur une face et « SL « sur l'autre : boite de 30 comprimés sous plaquettes thermoformées en PYDC/aluminium.
COMPOSITION
Comprimés dosés à 400 mg d'imatinib (sous forme de mésilate)
Excipients communs: Noyau du comprimé : cellulose microcristalline, crospovidone, hypromellose, stéarate de magnésium, silice colloïdale anhydre.Pelliculage du comprimé : oxyde de fer rouge (E172), oxyde de fer jaune (E172), macrogol.talc, hypromellose.
CLASSE PHARMACEUTIQUE
Inhibiteur de protéine-tyrosine kinase.
INDICATIONS
Glivec est indiqué dans le traitement :des patients adultes et enfants atteints de leucémie myéloïde chronique (LMC) chromosome Philadelphie (bcr-abl) positive (Ph+) nouvellement diagnostiquée lorsque la greffe de moelle osseuse ne peut être envisagée comme un traitement de première intention (cf. Posologie et mode d'administration pour l'utilisation chez l'enfant).des patients atteints de LMC Ph+ en phase chronique après échec du traitement par l'interféron alpha, ou en phase accélérée ou en crise blastique (cf. Posologie et mode d'administration pour l'utilisation chez l'enfant).L'effet de Glivec sur l'issue d'une greffe de moelle osseuse n'a pas été évalué.Glivec est également indiqué dans le traitement des patients adultes atteints de tumeurs stromales gastro-intestinales (GIST - gastrointestinal stromal tumours) malignes Kit (CD 117) positives non résécables et/ou métastatiques.
Chez l'adulte, l'efficacité de Glivec est basée sur les taux de réponses hématologiques et cytogénétiques globales et la survie sans progression dans la LMC et sur les taux de réponses objectives des patients adultes dans les GIST. Pour ces 2 maladies, il n'existe pas d'étude clinique contrôlée démontrant un bénéfice clinique ou une prolongation de la durée de vie.
POSOLOGIE ET MODE D'ADMINISTRATION
La dose prescrite doit être administrée par voie orale avec un grand verre d'eau, au cours d'un repas pour réduire le risque d'irritations gastrointestinales. Les doses de 400 mg ou 600 mg devront être administrées en une prise par jour, tandis que la dose journalière de 800 mg devra être répartie en deux prises de 400 mg par jour, matin et soir.
Pour les patients incapables d'avaler les comprimés pelliculés, il est possible de disperser ces comprimés dans un verre d'eau minérale ou de jus de pomme. Le nombre de comprimés requis devra être placé dans un volume de boisson approprié (approximativement 200 ml pour un comprimé à 400 mg) et être remué avec une cuillère. La suspension devra être administrée immédiatement après désagrégation complète du (des) comprimés.
- Posologie dans la LMC chez l'adulte
• Patients en phase chronique de LMC : la posologie recommandée est de 400 mg/j. La phase chronique est définie par l'ensemble des critères suivants : blastes < 15 % dans le sang et la moelle osseuse, basophiles dans le sang < 20 %, plaquettes > 100 x 10°/l.
• Patients en phase accélérée : la posologie recommandée est de 600 mg/j. La phase accélérée est définie par la présence d'un des critères suivants : blastes > 15 % mais < 30 % dans le sang ou la moelle osseuse, blastes plus promyélocytes > 30 % dans le sang ou la moelle osseuse (à condition que blastes < 30 %), basophiles dans le sang 2 20 %, plaquettes < 100 x 10c/l indépendamment du traitement.
Patients en crise blastique : la posologie recommandée est de 600 mg/j. La crise blastique est définie par la présence de blastes > 30 % dans le sang ou la moelle osseuse ou un envahissement extramédullaire autre qu'une hépatosplénomégalie.
Durée du traitement : dans les études cliniques, le traitement est poursuivi jusqu'à progression de la maladie. L'effet de l'arrêt du traitement après l'obtention d'une réponse cytogénétique complète n'a pas été étudié. En l'absence d'effets indésirables sévères et de neutropénie ou de thrombopénie sévères non imputables à la leucémie, une augmentation de la dose peut être envisagée, de 400 mg à 600 mg ou 800 mg, chez les patients en phase chronique, ou de 600 mg à un maximum de 800 mg (en deux prises de 400 mg par jour) chez les patients en phase accélérée ou en crise blastique, dans les circonstances suivantes : évolution de la maladie (à tout moment), absence de réponse hématologique satisfaisante après un minimum de 3 mois de traitement, absence de réponse cytogénétique après 12 mois de traitement, ou perte de la réponse hématologique et/ou cytogénétique obtenue auparavant. Les patients devront être surveillés étroitement après augmentation de la dose étant donnée la possibilité d'une incidence accrue des effets indésirables à plus fortes doses.
- Posologie dans la LMC chez l'enfant :
Chez l'enfant, la posologie devra être établie en fonction de la surface corporelle (mg/m2). La dose journalière recommandée chez l'enfant est de 340 mg/m2 dans la LMC en phase chronique et dans la LMC en phase avancée (ne doit pas dépasser une dose totale de 800 mg). Le traitement peut être administré en une prise quotidienne ou bien être divisé en deux prises (une le matin et une le soir). Ces recommandations posologiques reposent actuellement sur un faible nombre d'enfants (cf. Propriétés pharmacodynamiques et Propriétés pharmacocinétiques). On ne dispose d'aucune donnée chez l'enfant de moins de 2 ans. L'augmentation de doses de 340 mg/m2 jusqu'à 570 mg/m2 par jour (sans dépasser la dose totale de 800 mg) peut être envisagée chez l'enfant en l'absence d'effets indésirables graves et de neutropénie ou thrombopénie sévères non liées à la leucémie dans les circonstances suivantes : progression de la maladie (à n'importe quel moment) ; absence de réponse hématologique satisfaisante après au moins 3 mois de traitement ; absence de réponse cytogénétique après 12 mois de traitement ; ou perte d'une réponse hématologique et/ou cytogénétique antérieure. Les patients devront être surveillés attentivement au cours des escalades de doses compte tenu du risque accru d'effets indésirables à des doses plus élevées.
- Posologie dans les LAL Ph+
La posologie recommandée de Glivec est de 600 mg/ jour chez les patients atteints de LAL Ph+. Le traitement devrait être supervisé par des hématologues experts dans la prise en charge de cette maladie pour toutes les phases de traitement.Schéma thérapeutique : Sur la base des données existantes, Glivec s'est montré efficace et sûre lorsqu'il est administré à 600 mg/j en association à une chimiothérapie d'induction, de consolidation et d'entretien utilisée des LAL Ph+ nouvellement diagnostiquées de l'adulte (cf. Propriétés pharmacodynamiques). La durée de traitement par Glivec peut varier en fonction du traitement appliqué, mais généralement les traitements prolongés de Glivec ont fourni de meilleurs résultats.
Chez les patients adultes atteints de LAL Ph+ en rechute ou réfractaire, une monothérapie par Glivec à la dose de 600 mg/j est sure, efficace et peut être poursuivie jusqu'à la progression de la maladie.
- Posologie dans les GIST
Patients atteints de GIST malignes non résécables et/ou métastatiques : la posologie recommandée est de 400 mg/j. Il existe des données limitées concernant l'effet de l'augmentation des doses de 400 mg à 600 mg ou 800mg chez des patients en progression lorsqu'ils sont traités à la plus faible dose (cf. Propriétés pharmacodynamiques). Il n'y a pas actuellement de données disponibles permettant de recommander une dose spécifique en fonction de la résection gastro-intestinale antérieure chez des patients atteints de GIST (cf. Propriétés pharmacocinétiques). La majorité des patients (98 %) inclus dans l'étude clinique avaient eu une résection auparavant. Pour tous les patients de l'étude, la première administration de Glivec a eu lieu au moins deux semaines après la résection; toutefois on ne peut pas faire d'autre recommandation supplémentaire sur la base de cette étude. Durée du traitement : dans les études cliniques menées chez des patients atteints de GIST, le traitement par Glivec a été poursuivi jusqu'à la progression de la maladie. A la date de l'analyse, la durée médiane de traitement était de 7 mois (7 jours à 13 mois). L'effet de l'arrêt du traitement après l'obtention d'une réponse n'a pas été étudié.
- Posologie dans le DFSP
La posologie recommandée de Glivec est de 800 mg/jour chez les patients atteints de DFSP.
- Ajustement de la posologie en cas d'effets indésirables
• Effets indésirables extra-hématologiques : En cas de survenue d'un effet indésirable extra-hématologique sévère, le traitement par Glivec doit être interrompu jusqu'à résolution de l'événement. Le traitement peut ensuite être repris de manière appropriée en fonction de la sévérité initiale de l'événement. En cas d'élévation de la bilirubine > 3 fois la limite supérieure de la normale (LSN) fournie par le laboratoire d'analyses ou des transaminases > 5 fois la LSN, Glivec doit être interrompu jusqu'à un retour de la bilirubine à un taux < 1,5 fois la LSN et des transaminases à un taux < 2,5 fois la LSN. Le traitement peut alors être repris à dose réduite chez l'adulte, la dose sera diminuée de 400 à 300 mg ou de 600 à 400 mg ou de 800 mg à 600 mg, et chez l'enfant la dose sera diminuée de 340 à 260 mg/m2/jour.
• Effets indésirables hématologiques : En cas de neutropénie ou thrombopénie sévères, il est recommandé de diminuer la dose ou d'interrompre le traitement conformément au tableau ci-dessous. Ajustements de posologie en cas de neutropénie et de thrombocytopénie 
• Enfant : Il n'y a pas d'expérience chez l'enfant de moins de 2 ans (cf. Propriétés pharmacodynamlques). L'utilisation chez l'enfant atteint de LAL Ph+ est limitée. Il n'y a pas d'expérience chez l'enfant et l'adolescent atteints de GIST. Insuffisance hépatique .: L'imatinib est principalement métabolisé par le foie. Les patients présentant une altération de la fonction hépatique, légère, modérée ou importante devraient être traités à la dose minimale recommandée de 400 mg. La dose peut être réduite si le patient développe une toxicité inacceptable (cf. Mises en garde spéciales et précautions particulières d'emploi, Effets indésirables et Propriétés pharmacocinétiques).
Classification des altérations hépatiques
| Altération de la fonction hépatique
|
Tests de la fonction hépatique |
| Légère
|
Augmentation de la bilirubine totale de 1,5 fois la LSN ASAT > LSN (peut être normale ou < LSN si la bilirubine totale est > LSN) |
| Modérée
|
Augmentation de la bilirubine totale > 1,5 fois la LSN et < 3,0 fois la LSN ASAT quelle que soit la valeur. |
| Importante
|
Augmentation de la bilirubine totale > 3,0 fois la LSN et < 10 fois la LSN ASAT quelle que soit la valeur. |
LSN : Limite supérieure de la normale du laboratoire ASAT : aspartate aminotransférase
• Insuffisance rénale : aucune étude clinique n'a été conduite chez des patients présentant une altération de la fonction rénale (les patients avec une créatininémie supérieure à 2 fois la limite supérieure normale étaient e/clus des études). Cependant, l'imatinib et ses métabolites n'étant pas excrétés par le rein de façon significative et la clairance rénale de l'imatinib étant négligeable, une diminution de la clairance totale n'est pas attendue chez les patients ayant une insuffisance rénale. Toutefois, la prudence est recommandée chez les patients présentant une insuffisance rénale sévère.
• Patients âgés : la pharmacocinétique de l'imatinib n'a pas été spécifiquement étudiée chez le sujet âgé. Aucune différence significative de pharmacocinétique n'a été observée en fonction de l'âge chez les patients adultes inclus dans les études cliniques dont plus de 20 % étaient âgés de 65 ans et plus. Par conséquent, aucune recommandation particulière sur la posologie n'est requise pour ces patients.
CONTRE-INDICATIONS
Hypersensibilité à la substance active ou à l'un des excipients.
MISES EN GARDE et PRÉCAUTIONS D'EMPLOI
Lorsque Glivec est co-administré avec d'autres médicaments, des interactions médicamenteuses sont possibles (cf. Interactions avec d'autres médicaments et autres formes d'interaction). L'utilisation concomitante d'imatinib et de médicaments inducteurs du CYP3A4 [par exemple: dexaméthasone, phénytoïne, carbamazépine, rifampicine, phénobarbital, ou Hypericum perforatum (millepertuis)], peut réduire signiflcativement l'exposition systémique au Glivec et augmenter potentiellement le risque d'échec thérapeutique. Par conséquent, l'utilisation concomitante d'imatinib avec des inducteurs puissants du CYP3A4 devra être évitée (cf. Interactions avec d'autres médicaments et autres formes d'interaction). Le métabolisme de Glivec est principalement hépatique, et seulement 13 % de l'excrétion est rénale. Chez les patients présentant une altération de la fonction hépatique (légère, modérée ou importante) la numération formule sanguine et les enzymes hépatiques devront être étroitement surveillées (cf. Posologie et mode d'administration, Effets indésirables et Propriétés pharmacocinétiques). Il est à noter que lés patients atteints de GIST peuvent présenter des métastases hépatiques qui pourraient entraîner une insuffisance hépatique. Lorsque l'imatinib est associé à des chimiothérapies à fortes doses chez des patients atteints de LAL Ph+, une toxicité hépatique transitoire se traduisant par une élévation des transaminases et une hyperbilirubinémie a pu être observee. La surveillance étroite de la fonction hépatique devra être prise en compte lorsque l'imatinib est associé à une chimiothérapie connue comme pouvant être associée à une altération de la fonction hépatique (cf. Interactions avec d'autres médicaments et autres formes d'interaction et Effets indésirables). Des cas de rétention hydrique sévère (épanchement pleural, oedème, oedème pulmonaire, ascite) ont été décrits chez environ 1 à 2 % des patients traités. Il est donc fortement recommandé de peser régulièrement les patients. Une prise de poids rapide et inattendue devra faire l'objet d'un examen plus approfondi et, si nécessaire, l'instauration d'un traitement symptomatique et des mesures thérapeutiques devront être entreprises. Dans les études cliniques, l'incidence de ces effets indésirables était augmentée chez les patients âgés ainsi que chez ceux ayant des antécédents cardiaques. La prudence est donc recommandée chez des patients présentant un dysfonctionnement cardiaque. Dans l'étude clinique menée dans les GIST, des hémorragies gastro-intestinales et intratumorales ont été rapportées (cf. Effets indésirables). Sur la base des données disponibles, aucun facteur (ex. taille de la tumeur, localisation de la tumeur, troubles de la coagulation) n'a été identifié, prédisposant les patients atteints de GIST à un risque plus élevé de développer l'un ou l'autre des deux types d'hémorragies. Puisqu'une augmentation de la vascularisation et une propension aux saignements font partie de la nature et l'évolution clinique de la maladie, des modalités standardisées de suivi et de prise en charge des hémorragies devront être adoptées pour tous les patients.
Analyses biologiques. Des numérations formules sanguines doivent être effectuées régulièrement : le traitement par Glivec de patients atteints de LMC a été associé à une neutropénie ou une thrombopénie. Cependant, la survenue de ces cytopénies est vraisemblablement liée au stade de la maladie traitée : elles ont été plus fréquemment retrouvées chez les patients en phase accélérée ou en crise blastique que chez ceux en phase chronique de la LMC. Le traitement peut alors être interrompu ou la dose réduite, selon les recommandations de la rubrique Posologie et mode d'administration. La fonction hépatique (transaminases, bilirubine, phosphatases alcalines) doit faire l'objet d'une surveillance régulière.Glivec et ses métabolites ne sont pas excrétés de façon significative par le rein (13 %) et bien que ia clairance de la créatinine diminue avec l'âge, l'âge n'altère pas de façon significative les paramètres pharmacocinétiques de Glivec.
CONDUITE ET UTILISATION DE MACHINES
Les effets sur l'aptitude à conduire des véhicules et à utiliser des machines n'ont pas été étudiés. Toutefois, les patients devront être informés qu'ils peuvent voir survenir des effets indésirables tels que sensations vertigineuses, troubles visuels au cours du traitement par l'imatinib. La prudence est donc recommandée pour les utilisateurs de véhicules ou de machines.
INTERACTIONS
Substances actives pouvant augmenter les concentrations plasmatiques d'imatinib Les substances inhibant l'activité de l'isoenzyme CYP3A4 du cytochrome P450 (par exemple : kétoconazole, itraconazole, érythromycine, clarithromycine) pourraient diminuer le métabolisme d'imatinib et donc augmenter les concentrations plasmatiques de l'imatinib. Une augmentation significative de l'exposition systémique à l'imatinib (la valeur moyenne de la Cma(et de l'ASC (Aire sous la courbe) ont respectivement été augmentées de 26 % et 40 %) a été observée chez des volontaires sains lors de l'administration d'une dose unique de kétoconazole (un inhibiteur du CYP3A4). La prudence est requise lorsque Glivec est administré avec des inhibiteurs du CYP3A4.
• Substances actives pouvant diminuer les concentrations plasmatiques d'imatinib : Les substances agissant comme inducteurs de l'activité du CYP3A4 pourraient augmenter le métabolisme de l'imatinib et diminuer ses concentrations plasmatiques. Les traitements concomitants induisant le CYP3A4 [par exemple : dexaméthasone, phénytoïne, carbamazépine, rifampicine, phénobarbital, Hypericum perforatum (millepertuis)] pourraient donc réduire significativement l'exposition systémique au Glivec, et potentiellement augmenter le risque d'échec thérapeutique. Un traitement préalable par 600 mg de rifampicine à doses multiples suivies d'une dose unique de 400 mg de Glivec, a entraîné une diminution de CmM et de l'ASC(M4) d'au moins 54 % et 74 %, par rapport à leurs valeurs respectives sans traitement par rifampicine L'utilisation concomitante d'imatinib avec des inducteurs puissants du CYP3A4 devra être évitée.
• Substances actives dont la concentration plasmatique peut être modifiée par Glivec : L'imatinib augmente la valeur moyenne de la CmB/ et l'ASC de la simvastatine (substrat du CYP3A4), respectivement 2 fois et 3,5 fois, indiquant ainsi une inhibition du CYP3A4 par l'imatinib. Glivec doit donc être associé avec prudence à des substrats du CYP3A4 dont l'index thérapeutique est étroit (par exemple : ciclosporine ou pimozide). Par ailleurs, Glivec peut augmenter la concentration plasmatique d'autres médicaments métabolisés par le CYP3A4 (par exemple triazolo-benzodiazépines, inhibiteurs calciques de type dihydropyridine, certains inhibiteurs de l'HMG-CoA réductase, c'est-à-dire les statines, etc.).
La warfarine étant métabolisée par le CYP2C9, les patients nécessitant un traitement anticoagulant devront recevoir de l'héparine standard ou de bas poids moléculaire.In vitro, Glivec inhibe l'activité de l'isoenzyme CYP2D6 du cytochrome P450 à des concentrations similaires à celles affectant l'activité du CYP3A4. L'exposition systémique aux substrats du CYP2D6 est donc potentiellement augmentée lorsque Glivec est co-administré. Par précaution, en l'absence d'étude clinique, la prudence est donc recommandée.
In vitro, Glivec inhibe l'O-glycuroconjugaison du paracétamol (Ki = 58,5 mol/l aux doses thérapeutiques).La prudence est donc requise lors de l'utilisation concomitante de Glivec et de paracétamol, en particulier, avec de fortes doses de paracétamol.
Chez les patients atteints de LAL Ph+, on dispose d'une expérience clinique de l'administration concomitante de Glivec avec une chimiothérapie (cf. Propriétés pharmacodynamiques), cependant les interactions médicamenteuses entre l'imatinib et les schémas thérapeutiques de chimiothérapie n'ont pas été clairement identifiés. Les effets indésirables de l'imatinib tels qu'une hépatotoxicité, une myélosuppression ou d'autres effets, peuvent augmenter et il a été rapporté qu'une utilisation concomitante de la L-asparaginase pourrait être associée à une hépatotoxicité augmentée (cf. Effets indésirables). Ainsi, l'administration de Glivec en association nécessite des précautions particulières.
GROSSESSE et ALLAITEMENT
GROSSESSE
II n'existe pas de données suffisamment pertinentes concernant l'utilisation de l'imatinib chez la femme enceinte. Des études effectuées chez l'animal ont toutefois mis en évidence une toxicité sur la reproduction (cf. Données de sécurité précliniques) et le risque potentiel sur le foetus en clinique n'est pas connu. Glivec ne doit pas être utilisé à moins d'une nécessité absolue. S'il est utilisé au cours de la grossesse, la patiente doit être prévenue du risque potentiel pour le foetus. Chez les femmes en âge de procréer, une contraception efficace doit être conseillée pendant le traitement.
ALLAITEMENT
Chez l'animal, l'imatinib et/ou ses métabolites sont largement excrétés dans le lait. Par précaution, en l'absence de données cliniques, les femmes traitées par Glivec ne devraient pas allaiter.
EFFETS INDÉSIRABLES
Les patients atteints de pathologies malignes à un stade avancé peuvent présenter des affections intercurrentes. Ces affections peuvent rendre difficile l'évaluation du lien de causalité à Glivec des événements indésirables en raison de la variété des symptômes liés à la maladie sous-jacente, à sa progression ou à la co-administration de nombreux médicaments.Au cours des études cliniques menées dans la LMC, un arrêt du traitement motivé par des effets indésirables imputables au médicament a été observé chez 2% des patients nouvellement diagnostiqués, 4% des patients en phase chronique tardive après échec du traitement par l'interféron, 4% des patients en phase accélérée après échec du traitement par l'interféron et 5% des patients en crise blastique après échec du traitement par l'interféron. Dans les GIST, le produit étudié a été arrêté en raison d'effets indésirables imputables au médicament chez 4% des patients.
Les effets indésirables ont été comparables dans toutes les indications, à deux exceptions près. Il y a eu plus de myélosuppressions observées chez les patients atteints de LMC que chez ceux atteints de GIST, ce qui est probablement dû à la maladie sous-jacente. Dans l'étude clinique GIST, 7 (5 %) patients ont présenté des saignements de grade 3 / 4 selon la classification CTC (Common Toxicity Criteria) : saignements gastrointestinaux (3 patients), saignements intra-tumoraux (3 patients), les deux types (1 patient). La localisation de la tumeur gastro-intestinale peut avoir été à l'origine des saignements gastro¬intestinaux (cf. Mises en garde spéciales et précautions particulières d'emploi). Les saignements gastro-intestinaux et intra-tumoraux peuvent être sérieux et dans certains cas fatals.
Les effets indésirables les plus fréquemment rapportés (≥ 10 %) pouvant être imputables au traitement par Glivec dans les deux indications ont été des nausées modérées, vomissements, diarrhée, douleur abdominale, fatigue, myalgies, crampes musculaires et rash. Des oedèmes superficiels ont été très fréquemment observés dans toutes les études cliniques et décrits principalement comme des oedèmes périorbitaux ou des membres inférieurs.
Toutefois, ces oedèmes ont été rarement sévères et ont pu être contrôlés par des diurétiques, d'autres mesures symptomatiques ou en réduisant la dose de Glivec. Lorsque l'imatinib était associé à des doses élevées de chimiothérapie chez des patients atteints de LAL Ph+, une toxicité hépatique transitoire se traduisant par une élévation des transaminases et une hyperbilirubinémie a été observée. Divers effets indésirables tels qu'épanchement pleural, ascite, oedème pulmonaire, prise de poids rapide avec ou sans oedème superficiel ont été décrits dans le cadre de rétention hydrique. Ces effets peuvent habituellement être contrôlés par l'interruption temporaire du Glivec et par l'utilisation de diurétiques et d'autres traitements symptomatiques appropriés. Cependant, certains de ces effets peuvent être graves voire mettre enjeu le pronostic vital : plusieurs patients en crise blastique sont décédés, avec un tableau clinique complexe associant un épanchement pleural, une insuffisance cardiaque congestive et une insuffisance rénale. Les études cliniques menées chez l'enfant n'ont pas révélé de données de tolérance particulière à cette population.
Les effets indésirables
Les effets indésirables, en dehors des cas isolés, sont repris ci-dessous : ils sont classés par organe et par ordre de fréquence en utilisant la convention suivante : très fréquent (>1/10), fréquent (>1/100, £1/10), peu fréquent (>1/1000, <1/100), rare <1/1000). Au sein de chaque fréquence de groupe, les effets indésirables doivent être présentés suivant un ordre décroissant de gravité. 
- Anomalies biologiques :
Paramètres hématologiques Dans les LMC, des cytopénies, en particulier des neutropénies et des thrombopénies, ont été régulièrement rapportées dans toutes es études cliniques avec une fréquence plus élevée au/ fortes doses ^ 750 mg (étude de phase I. Cependant, la survenue des cytopénies dépendait aussi nettement du stade de la maladie : la fréquence des neutropénies et des thrombopénies de grade 3 ou 4 (PN < 1,0 x 109/l ; taux de plaquettes < 50 x 109/l) étant 4 à 6 fois plus élevée dans les LMC en crise blastique ou en phase accélérée (respectivement 59-64% pour les neutropénies et 44-63% pour les thrombopénies) que dans les LMC en phase ironique nouvellement diagnostiquées (15% de neutropénie et 8,5% de thrombopénie). Dans les UVIC en phase chronique nouvellement diagnostiquées, les neutropénies et les thrombopénies de grade 4 (PN< 0,5 x 109/l ; plaquettes < 10 / 109/l) ont été observées chez 3% et < 1% des patients respectivement. La durée médiane des épisodes de neutropénie est habituellement de l'ordre de 2 à 3 semaines, et de 3 à 4 semaines pour les épisodes de thrombopénie. Ces événements peuvent être j habituellement contrôlés soit par une réduction de la dose soit par une interruption du traitement par Glivec, mais peuvent dans de rares cas conduire à une interruption définitive du traitement.
En pédiatrie, chez les enfants atteints de LMC, les toxicités les PLUS fréquemment observées étaient des cytopénies de grade 3 ou 4 incluant des neutropénies, des thrombocytopénies et des anémies. Elles surviennent principalement dans les premiers mois de traitement.
Chez les patients atteints de GIST, une anémie de grade 3 ou 4 a été rapportée respectivement chez 5,4% et 0,7% des patients. Ces cas d'anémies pouvaient être liés à un saignement gastro-intestinal ou Intra-tumoral, au moins chez certains de ces patients. Des neutropénies de grade 3 ou 4 ont été apportées respectivement chez 7,5% et 2,7% des patients et une thrombopénie de grade 3 chez 0,7% des patients.
Aucun patient n'a développé de thrombopénie de grade 4. Des diminutions du nombre de leucocytes et de neutrophiles ont principalement été observées au cours des six premières semaines du traitement, les valeurs demeurant relativement stables par la suite.
• Paramètres biochimiques
Des augmentations importantes des transaminases ou de la bilirubine ont été peu fréquentes (< 3% des patients atteints de LMC) et ont été habituellement contrôlées par une réduction de la dose ou une interruption du traitement (la durée médiane de ces épisodes était d'environ une semaine). Le traitement a été interrompu définitivement en raison : anomalies biologiques hépatiques chez moins de 0,5% des patients atteints de LMC.
Chez les patients atteints de GIST (étude B2222) on a observé 6,8% d'augmentations de grade 3 à 4 des ALAT (alanine aminotransférase) et 4,8% d'augmentations de grade 3 à 4 des ASAT (aspartate aminotransférase). Une augmentation de la bilirubine était inférieure à 3% des patients. Il y a eu des cas d'hépatite cytolytique et cholestatique et de défaillance hépatique, dont certains avec une issue fatale, y compris un patient sous dose élevée de paracétamol.
SURDOSAGE
L'expérience de l'utilisation de doses supérieures à 800 mg est limitée. Des cas isolés de surdosage avec Glivec ont été rapportés. Un patient en crise blastique myéloïde ayant pris, par inadvertance, Glivec à la dose de 1200 mg pendant 6jours a présenté une élévation de la créatininémie de grade 1, une ascite, une élévation des transaminases hépatiques de grade 2 et une élévation de la bilirubine de grade 3. Le traitement a été interrompu temporairement et toutes les anomalies se sont normalisées en 1 semaine. Le traitement a pu être repris à la close de 400 mg sans récidive des problèmes. Un autre patient a développé des crampes musculaires sévères après avoir pris Glivec à la dose de 1600 mg par jour pendant 6 jours. A la suite de l'interruption du traitement, une résolution complète des crampes musculaires a été obtenue et le traitement a pu être repris. En cas de surdosage, le patient devra être placé sous surveillance et recevoir un traitement symptomatique approprié.
PHARMACODYNAMIE
L'imatinib est un inhibiteur de protéine tyrosine kinase qui inhibe aissamment la tyrosine kinase Bcr-Abl au niveau cellulaire in vitro et in vivo. Le produit inhibe sélectivement la prolifération et induit une apoptose dans les lignées cellulaires Bcr-Abl positives ainsi que dans les cellules leucémiques fraîches provenant de patients atteints de LMC ou de leucémie aiguë lymphoblastique (LAL) chromosome Philadelphie positives, in vivo, le produit présente une activité anti-tumorale lorsqu'il est administré en monothérapie chez l'animal porteur de cellules tumorales Bcr-Abl positives. L'imatinib est également un inhibiteur des tyrosines kinases du récepteur du PDGF (platelet-derived growth factor), PDGF-R et du SCF (stem cell factor) c-Kit et il inhibe les processus cellulaires médiés par le PDGF et le SCF.
In vitro, l'imatinib inhibe la prolifération et induit une apoptose des cellules de tumeur stromale gastro-intestinale (GIST), qui expriment une mutation activatrice du kit. L'activation constitutive du récepteur du PDGF ou des tyrosine kinases Abl, résultant de la fusion à différentes protéines partenaires ou de la production constitutive de PDGF, sont impliquées dans la pathogénèse du DFSP. L'imatinib inhibe la signalisation et la prolifération des cellules sensibles à l'activité dérégulée des kinases Abl ou PDGFR.
- Etudes cliniques dans la leucémie myéloïde chronique
L'efficacité de Glivec est basée sur les taux de réponses cytogénétiques et hématologiques globales. Il n'existe pas actuellement d'étude contrôlée démontrant un bénéfice clinique tel qu'une amélioration des symptômes liés à la maladie ou une prolongation de la durée de vie.
Trois grandes études ouvertes internationales de phase II, non contrôlées, ont été menées chez des patients atteints de LMC chromosome Philadelphie positive (Ph+) en phase avancée, crise blastique ou phase accélérée, ainsi que chez des patients atteints d'autres leucémies Ph+ ou de LMC en phase chronique en échec d'un traitement antérieur par Interféron alpha (IFN). Une vaste étude randomisée ouverte, multicentrique, internationale de phase III a été menée chez des patients présentant une LMC Ph+ nouvellement diagnostiquée. De plus, des enfants ont été traités dans deux études de phase I et une étude de phase II. Dans toutes les études cliniques, 38-40 % des patients avaient -50 ans et 10-12 % des patients avaient à 70 ans.
• Phase chronique nouvellement diagnostiquée : Cette étude de phase III, chez les patients adultes a comparé l'administration de Glivec en monothérapie à une association d'interféron-alpha (IFN) et de cytarabine (Ara-C). Les patients présentant une absence de réponse [absence de réponse hématologique complète (RHC) à 6 mois, augmentation des leucocytes, absence de réponse cytogénétique majeure (RCyM) à 24 mois], une perte de la réponse de la RHC ou de la RCyM) ou une intolérance sévère au traitement ont été autorisés à permuter dans l'autre groupe de traitement. Dans le groupe recevant Glivec, les patients ont été traités par 400 mg/jour. Dans le groupe recevant IFN, les patients ont reçu une dose cible d'IFN de 5 MUI/m2/jour par voie sous-cutanée, en association à une dose sous-cutanée d'Ara-C de 20 mg/m2/jour pendant 10 jours/mois. Au total, 1106 patients ont été randomisés, soit 553 patients par groupe de traitement.
es caractéristiques initiales étaient bien équilibrées dans les deux groupes de traitement. L'âge médian était de 51 ans (extrêmes : 18-70 ans) dont 21,9% des patients âgés de 60 ans ou plus. Les groupes étaient composés de 59% d'hommes et 41% de |'extrêmes, de 89,9% de blancs et 4,7% de noirs. Le suivi médian de tous les patients était respectivement de 31 et 30 mois dans les groupes traités par Glivec et par l'IFN. Dans cette étude, le paramètre d'évaluation principal de l'efficacité est la survie sans agression de la maladie.
La progression était définie comme l'un des événements suivants : évolution vers une phase accélérée ou une crise blastique, décès, perte de la RHC ou de la RCyM, ou, chez les patients n'obtenant pas une RHC, augmentation des leucocytes malgré une prise en charge thérapeutique adaptée. La réponse cytogénétique majeure, la réponse hématologique, la réponse moléculaire (évaluation de la maladie résiduelle), le délai avant apparition d'une phase accélérée ou d'une crise blastique et la survie sont les principaux paramètres d'évaluation secondaires.
Le Tableau 1 présente les données relatives au/ types de réponses. La réponse hématologique complète, la réponse cytogénétique majeure et la réponse cytogénétique complète aussi bien que la réponse moléculaire majeure ont été significativement plus élevées dans le groupe Glivec par comparaison au groupe IFN + Ara-C. Avec les données de suivi actuellement disponibles, le taux estimé de patients sans évolution vers une phase accélérée ou une crise blastique à 30 mois est significativement plus élevé avec Glivec par rapport à l'IFN (94,8% versus 89,6%, p<0,0016). Le tau/ estimé de survie sans progression à 30 mois était de 87,8% avec Glivec, contre 68,3% dans le groupe témoin. Il y a eu respectivement 33 et 46 décès rapportés dans les groupes Glivec et IFN, avec un taux de survie estimé à 30 mois de 94,6% et 91,6% respectivement (différence non significative). La probabilité de survie sans progression à 30 mois était de 100% pour les patients qui ont présenté une réponse cytogénétique complète avec une réponse moléculaire majeure (réduction d'au moins 3 log) à 12 mois par comparaison à 93% pour les patients qui ont présenté une réponse cytogénétique complète sans réponse moléculaire majeure et à 82% pour les patients qui n'ont pas présenté de réponse cytogénétique complète (p<0,001) à cette date. Dans cette étude, des augmentations de doses de 400 mg à 600 mg par jour, puis de 600 mg à 800 mg par jour ont été autorisées. Après 42 mois de suivi, une perte confirmée (dans les 4 semaines) de la réponse cytogénétique a été observée chez 11 patients. Parmi ces 11 patients, 4 patients ont eu une augmentation de dose jusqu'à 800 mg par jour et 2 d'entre eux ont obtenu de nouveau une réponse cytogénétique (1 partielle et 1 complète, cette dernière associée à une réponse moléculaire), tandis que parmi les 7 patients qui n'ont pas eu d'augmentation de doses, un seul patient a présenté de nouveau une réponse cytogénétique complète. Le pourcentage de certains effets indésirables observés était plus élevé chez les 40 patients dont la dose a été augmentée jusqu'à 800 mg par comparaison à la population de patients observée avant toute augmentation de la dose (n=551). Les effets indésirables les plus fréquents ont été des hémorragies gastrointestinales, des conjonctivites et une augmentation des transaminases ou de la bilirubine. D'autres effets indésirables ont été rapportés à des fréquences plus faibles ou équivalentes.
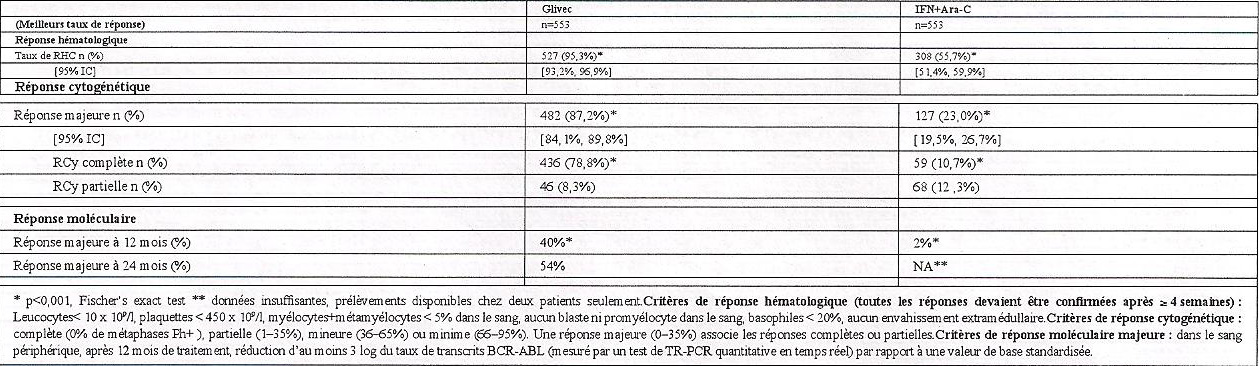
Tableau 1 : Réponses observées dans l'étude portant sur LMC de diagnostic récent (données à 30 mois)
• Phase chronique, échec de l'interféron : 532 patients adultes ont été traités avec une dose initiale de 400 mg. Les patients ont été répartis en trois catégories principales : échec hématologique (29 %), échec cytogénétique (35 %) ou intolérance à l'interféron (36%). Les patients avaient précédemment reçu un traitement par l'IFN à des doses > 25 x 106UI/semaine pendant une durée médiane de 14 mois et étaient tous en phase chronique tardive, avec un diagnostic établi depuis une durée médiane de 32 mois, Le critère principal d'efficacité de l'étude était le tau/ de réponse cytogénétique majeure (réponses complètes plus réponses partielles : 0 à 35 % de métaphases Ph+ dans la moelle osseuse).Dans cette étude, 65% des patients ont obtenu une réponse te que majeure, la réponse étant complète chez 53% (confirmée 43%) des patients (Tableau 2). Une réponse complète hématologique a été observée chez 95% des patients.
• Phase accélérée : L'étude a inclus 235 patients adultes en phase accélérée. Les 77 premiers patients ont commencé le traitement à 400 mg. Par la suite, le protocole a été amendé pour autoriser une posologie plus élevée et les 158 patients suivants ont commencé le traitement à 600 mg. Le critère principal d'efficacité était le taux de réponses hématologiques, défini comme une réponse complète hématologique, ou bien une disparition des signes de leucémie (c'est à dire : disparition des blastes de la moelle osseuse et du sang, mais sans la récupération hématologique périphérique totale observée dans le cas de réponse complète), ou encore un retour en phase chronique de la LMC. Une réponse hématologique confirmée a été obtenue chez 71,5% des patients (Tableau 2). Fait important, 27,7% des patients ont également présenté une réponse cytogénétique majeure, qui a été complète chez 20,4% des patients (confirmée 16%). Pour les patients ayant reçu une dose de 600 mg, les estimations actuelles de la médiane de survie sans progression et de la survie globale ont été de 22,9 mois et 42,5 MOIS respectivement.
Crise blastique myéloïde : 260 patients en crise blastique myéloïde ont été inclus. 95 patients (37 %) avaient reçu une chimiothérapie ("patients prétraités") comme traitement antérieur d'une phase accélérée ou d'une crise blastique p "55 (63 %) n'en avaient pas reçu ("patients non prétraités"). Les 37 premiers patients ont débuté le traitement à 400 mg. Le protocole a ensuite été amendé pour permettre une posologie plus élevée, et les 223 patients suivants ont reçu une dose initiale de 600 mg.
Le critère principal d'efficacité était le taux de réponse hématologique, défini comme soit une réponse complète hématologique, soit une disparition des signes de leucémie, soit un retour en phase chronique de la LMC, selon les mêmes critères que ceux de l'étude menée chez des patients en phase accélérée. Dans cette étude, 31 % des patients ont obtenu une réponse hématologique (36 % chez les patients non prétraités et 22 % chez les patients prétraités). Le taux de réponse a également été supérieur chez les patients traités par 600 mg (33 %) par rapport au/ patients traités par 400 mg (16%, p=0,0220). L'estimation actuelle de la médiane de survie des patients non prétraités est de 7,7 mois, contre 4,7 mois chez les patients prétraités.
• Crise blastique lymphoide : un nombre limité de patients ont été inclus dans les études de phase I (n=10). Le taux de réponse hématologique était de 70 % sur une durée de 2 à 3 mois.
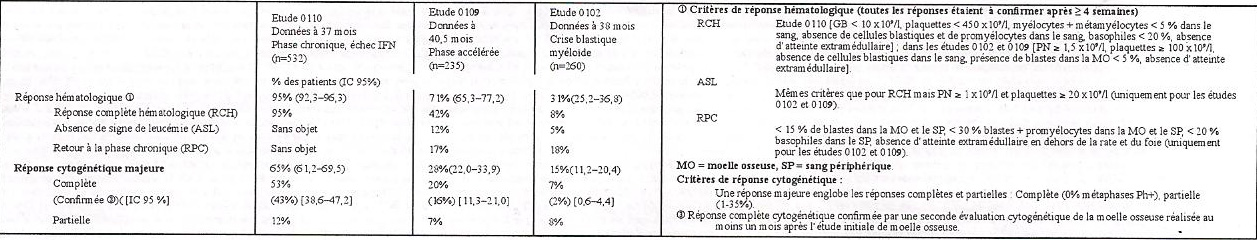
Tableau 2 : Réponses dans les études LMC chez les adultes
• Patients pédiatriques : Un total de 26 patients pédiatriques âgés de moins de 18 ans, présentant une LMC en phase chronique (n=11) ou une LMC en crise blastique ou une leucémie aiguë Ph+ (n=15), ont été recrutés dans une étude de phase I avec escalade de doses. Il s'agissait d'une population de patients lourdement prétraités, dans la mesure où 46 % avaient déjà bénéficié d'une transplantation médullaire et 73 % d'une polychimiothérapie. Les doses de Glivec administrées étaient de 260 mg/m2/jour (n=5), 340 mg/m2/jour (n=9), 440 mg/m2/jour (n=7) et 570 mg/m2/jour (n=5). Parmi les 9 patients atteints de LMC en phase chronique dont les données cytogénétiques sont disponibles, 4 (44 %) ont obtenu une réponse cytogénétique complète et 3 (33 %) une réponse cytogénétique partielle, pour un tau/ de RCyM de 77 %. Un total de 51 patients pédiatriques atteints d'une LMC en phase chronique nouvellement diagnostiquée non traitée ont été inclus dans une étude de phase II avec un seul bras, muticentrique en ouvert Ces enfants étaient traités par Glivec à la dose de 340 mg/m2/jour, sans interruption de traitement en l'absence de la dose limite toxique. Le traitement par Glivec induit une réponse rapide chez les patients pédiatriques atteints de LMC nouvellement diagnostiqués avec une RCH de 78% après 8 semaines de traitement. Le tau/ élevé de RCH s'accompagne d'une réponse complète cytogénétique de 65% qui est comparable au/ résultats observés chez les adultes. De plus, une réponse cytogénétique partielle était observée à un taux de 16% pour un tau/ de 81% de réponses cytogénétiques majeures. La majorité des patients qui ont atteint une réponse cytogénétique complète ont développé cette réponse entre 3 et 10 mois avec une estimation selon Kaplan-Meier de la durée médiane de réponse à 5,6 mois.
- Etudes cliniques dans la LAL Ph+ LAL Ph+ nouvellement diagnostiquée :
dans une étude contrôlée (ADE10) comparant l'imatinib versus chimiothérapie d'induction, chez 55 patients nouvellement diagnostiqués âgés de 55 ans et plus, l'imatinib utilisé seul a induit un tau/ signiflcativement plus élevé de réponse hématologique complète par rapport à la chimiothérapie (96,3% versus 50% ; p = 0,0001). Lorsque le traitement de rattrapage par l'imatinib a été administré aux patients qui n'avaient pas répondu ou avaient mal répondu à la chimiothérapie, 9 patients (81,8%) sur les 11 ont atteint une réponse hématologique complète. Cet effet clinique était associé à une plus forte réduction des taux de transcrits bcr-abl après deux semaines de traitement (p = 0,02) chez les patients traités par l'imatinib par rapport aux patients traités par chimiothérapie. Tous les patients ont reçu l'imatinib et une chimiothérapie de consolidation (voir Tableau 3) après le traitement d'induction et les taux de transcrits bcr-abl étaient identiques entre les deux bras après huit semaines de traitement. Comme on pouvait s'y attendre compte tenu du schéma de l'étude, aucune différence n'a été observée en termes de durée de rémission, de survie sans maladie ou de survie globale, même s'il faut noter que les patients en réponse moléculaire complète chez qui persistait une maladie résiduelle minime avait un pronostic plus favorable tant en termes de durée de rémission (p = 0,01) que de survie sans maladie (p = 0,02). Les résultats observés dans quatre études cliniques non contrôlées (AAU02, ADE04, AJP01 et AUS0I), dans une population de 211 patients atteints de LAL Ph+ nouvellement diagnostiquée, sont consistants avec les résultats décrits ci-dessus. L'imatinib en association avec la chimiothérapie d'induction (voir Tableau 3) a permis d'obtenir un tau/ de réponse complète hématologique de 93% (147 sur 158 patients évaluables) et un taux de réponse cytogénïque majeure de 90% (19 sur 21 patients évaluables). Le taux de réponse moléculaire complète était de 48% (49 sur 102 patients évaluables). La survie sans maladie et la survie globale ont constamment dépassé un an et elles étaient supérieures aux données historiques des groupes contrôles (survie sans maladie p<0,001 ; survie globale p<0,0001) dans les deux études (AJP01 et AUS0I).
Tableau 3 : Schémas des chimiothérapies utilisées en association avec l'imatinib 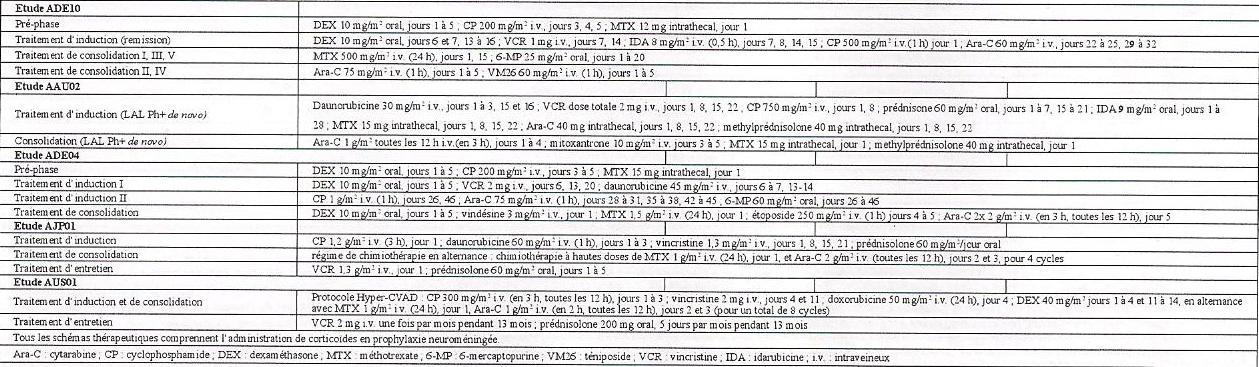
• LAL Ph+ en rechute ou réfractaire : Lorsque l'imatinib a été utilisé en monothérapie chez des patients atteints de LAL Ph+ en rechute ou réfractaire, il a été observé un taux de réponse hématologique de 30% (9% réponse complète) et un tau/ de réponse cryogénique majeure de 23% parmi les 53 patients évaluables pour la réponse sur un effectif total de 411 patients. (A noter que sur ces 411 patients, 353 avaient été traités dans le cadre d'un programme d'accès élargi au cours duquel la réponse primaire n'était pas collectée). La durée médiane jusqu'à la progression de la maladie dans la population globale de 411 patients atteints de LAL Ph+ en rechute ou réfractaire était de 2,6 à 3,1 mois, avec une médiane de survie globale allant de 4,9 à 9 mois chez 401 patients évaluables. Les données étaient identiques lorsque l'analyse a été de nouveau réalisée en prenant en compte uniquement les patients âgés de 55 ans et plus.
- Etudes cliniques dans les GIST
Une étude de Phase II, internationale, randomisée, en ouvert et non contrôlée a été conduite chez des patients atteints de tumeurs stromales gastro-intestinales (G IST) malignes non résécables ou métastatiques. Dans cette étude, 147 patients ont été inclus et randomisés pour recevoir soit 400 mg soit 600 mg de Glivec par jour par voie orale pour une durée pouvant atteindre 36 mois. Ces patients étaient âgés de 18 à 83 ans et ils avaient un diagnostic pathologique de G IST malignes Kit-positives, non résécables et/ou métastatiques. Le dosage immunohistochimique a été réalisé en routine avec des anticorps antiKit par (A-4502, antisérum polyclonal de lapin, 1:100 ; DAKO Corporation, Carpintería, CA) par la méthode utilisant le complexe peroxidase-biotine-avidine après marquage de l'antigène. Le critère principal d'efficacité était basé sur les taux de réponses objectives. Les tumeurs devaient être mesurables pour au moins un site de la maladie et la caractérisation de la réponse était basée sur les critères du Southwestern Oncology Group (SWOG). Les résultats sont présentés dans le Tableau 4.
Tableau 4 : Meilleure réponses tumorales dans l'essai STIB2222 (GIST)
| Meilleure réponses |
Toutes les doses (n=147) |
| 400 mg (n=73) |
|
| 600 mg (n=74) |
|
| n (%) |
|
| Réponse complète |
1 (0,7) |
| Réponse parallèle |
98 (66,7) |
| Stabilisation de la maladie |
23 (15,6) |
| Progression de la maladie |
18 (12,2) |
| Non évaluable |
5 (3,4) |
| Inconnu |
2 (1,4) |
Aucune différence des taux de réponses n'a été observée entre les deux groupes de dose. Un nombre important de patients qui présentaient une stabilisation de la maladie au moment de l'analyse intermédiaire a atteint une réponse partielle après une durée plus longue de traitement (médiane de suivi de 31 mois). La durée médiane pour obtenir une réponse était de 13 semaines (95%IC 12-23). La durée médiane jusqu'à échec du traitement chez les répondeurs était de 122 semaines (95%IC 106-147) alors que pour la population totale de l'étude, elle était de 84 semaines {95%IC 71-109). La durée médiane de survie globale n'a pas été atteinte. Après un suivi de 36 mois l'estimation selon Kaplan-Meier du tau/ de survie est de 68%. Dans les deux études cliniques (étude B2222 et une étude d'intergroupe S0033), la dose quotidienne de Glivec a été augmentée à 800 mg chez les patients qui progressaient au/ plus faibles doses de 400 mg ou 600 mg par jour. Au total 103 patients ont eu une augmentation de doses à 800 mg : 6 patients ont atteint une réponse partielle et 21 une stabilisation de leur maladie après augmentation de la dose pour un bénéfice clinique global de 26%. Sur la base des données de tolérance disponibles, l'augmentation de dose quotidienne à 800 mg chez des patients progressant au/ plus faibles doses de 400 mg ou 600 mg par jour ne semble pas affecter le profil de sécurité d'emploi de Glivec.
- Etudes cliniques dans le DFSP
Une étude ouverte multicentrique de phase II (étude B2225) a été menée incluant 12 patients atteints de DFSP traité par Glivec à 800 mg/jour. L'âge des patients atteints de DFSP allait de 23 à 75 ans ; leur maladie était métastatique ou en rechute locale après une chirurgie d'exérèse initiale et n'était pas considéré comme relevant d'une chirurgie d'exérèse supplémentaire au moment de l'entrée dans l'étude. La critère primaire d'efficacité reposait sur les tau/ de réponse objective. Parmi les 12 patients inclus, 9 ont répondu, 1 réponse complète et 8 réponses partielles. Trois des répondeurs partiels ont été rendu indemnes de maladie par chirurgie. La durée médiane de traitement dans l'étude B2225 était de 6,2 mois, avec une durée maximale de 24,3 mois. 6 autres patients atteints de DFSP et traités par Glivec ont été rapportés sous la forme de 5 observations individuelles, leur âge allait de 18 mois à 49 ans. Les patients adultes rapportés dans la littérature ont été traités par Glivec soit à la posologie de 400 mg/jour (4 cas) soit par 800 mg/jour (1 cas). Les enfants ont reçu une posologie de 400 mg/m2/j, augmenté par la suite à 520 mg/m2/j. 5 patients ont répondu, 3 complètement et 2 partiellement. La durée médiane de traitement dans la littérature allait de 4 semaines à plus de 20 mois. La translocation t(17:22) [(q22 :q13)], ou la protéine issue de ce gène hybride était présente chez pratiquement tous les répondeurs au traitement par Glivec.
PHARMACOCINETIQUE
Paramètres pharmacocinétiques de Glivec
La pharmacocinétique de Glivec a été évaluée à des doses comprises entre 25 et 1000 mg. Les profils pharmacocinétiques plasmatiques ont été analysés à J1, puis à J7 ou J28, au moment où les concentrations plasmatiques ont atteint un état d'équilibre.
• Absorption
La biodisponibilité absolue moyenne de l'imatinib est de 98 %. Il existe une forte variabilité inter-patient de l'ASC de l'imatimb plasmatique après une prise orale. Lorsqu'il est pris au cours d'un repas riche en lipides, le taux d'absorption de l'imatinib est peu réduit (diminution de 11 % de la C et prolongation de 1,5 h de t ), avec une légère diminution de l'ASC (7,4 %) comparée à une prise à jeun. L'effet d'une chirurgie gastro-intestinale antérieure sur l'absorption du produit n'a pas été étudié.
• Distribution
A des concentrations d'imatinib cliniquement significatives, la fraction liée au/ protéines plasmatiques est approximativement de 95 % sur la base des études in vitro, il s'agit principalement d'une liaison à l'albumine et au/ alpha-glycoprotéines acides, et dans une faible mesure au/ lipoprotéines.
• Métabolisme
Chez l'homme, le principal métabolite circulant est le dérivé piperazine N-demethyle qui présente in vitro une activité similaire à l'imatinib L'ASC plasmatique de ce métabolite n'atteint que 16% de l'ASC de l'imatinib. L'affinité pour les protéines plasmatiques du métabolite N-déméthylé est similaire à celle de la molécule mère. L'imatinib et le métabolite N-déméthylé représentent au total environ 65% du taux circulant de radioactivité (ASC(0-48h)). Le taux circulant de radioactivité restant correspond à un nombre de métabolites mineurs.
Les tests in vitro montrent que le CYP3A4 est le principal enzyme du cytochrome P450 humain catalysant la biotransformation de l'imatinib. Parmi un éventail de médicaments potentiellement co-administres (paracétamol, aciclovir, allopurinol, amphotericine, cytarabine, erythromycine fluconazole, hydroxyurée, norfloxacine, pénicilline Y) seuls l'érythromycine (IC50 50 µM) et le fluconazole (IC 118 |JM) ont montré une inhibition du métabolisme de l'imatinib pouvant être cliniquement significative. In vitro, l'imatinib est un inhibiteur compétitif des substrats marques du CYP2C9, des CYP2D6 et des CYP3A4/5 avec des valeurs de Ki de 27, 7,5 et 7,9 respectivement obtenues sur les microsomes hépatiques humains. Les concentrations plasmatiques maximales de l'imatinib sont de 2 -4 mol/1. Par conséquent une inhibition du métabolisme de produits co-administrés mettant enjeu les CYP2D6 et CYP3A4/5 est possible. L'imatinib n'interfère pas avec la biotransformation du 5-fluorouracile mais inhibe le métabolisme du paclitaxel par inhibition compétitive du CYP2C8 (K, = 34,7). Cette valeur de Ki est de loin supérieure au/ tau/ plasmatiques d'imatinib prévisibles chez les patients. Par conséquent, aucune interaction n'est attendue en cas de co-administration de l'imatinib avec le 5-fluorouracile ou le paclitaxel.
• Elimination
Apres administration d'une dose orale d'imatinib marqué au 14C, environ 81 % de la dose est éliminée au bout de 7 jours (68 % dans les fèces et 13 % dans les urines). La forme inchangée représente 25 % de la dose (5 % dans les urines, 20 % dans les fèces), le reste étant composé de métabolites.
- Pharmacocinétique plasmatique
Après administration par voie orale chez le volontaire sain, la demi-vie, d'environ 18 h, est compatible avec une prise quotidienne unique. L'augmentation de I ASC moyenne de l'imatinib est linéaire et proportionnelle à la dose administrée à des doses orales allant de 25 à 1000 mg. Lors d'administrations répétées en prise quotidienne unique, la cinétique de l'imatinib n'est pas modifiée, mais son accumulation à I état d équilibré, est augmentée d'un facteur de 1 5 à 2,5.
Pharmacocinétique chez des patients atteints de GIST Chez des patients atteints de GIST, l'e/position à l'état d'équilibre était 1,5 fois supérieure à celle observée à la même dose (400 mg/jour) chez des patients atteints de LMC Sur la base d'une analyse préliminaire de pharmacocinétique de population de patients atteints de GIST, on a identifié 3 variables (albumine, tau/ de globules blancs et bilirubine) qui présentaient une relation statistiquement significative avec la pharmacocinétique de l'imatinib. Des valeurs diminuées de l'albumine étaient associées à une diminution de la clairance (Cl/f) et les valeurs plus élevées du tau/ de globules blancs étaient associées a une réduction de la Cl/f. Toutefois, ces associations n'étaient pas suffisantes pour justifier un ajustement des doses. Dans cette population de patients, la présence de métastases hépatiques pourrait potentiellement conduire à une insuffisance hépatique et a une diminution du métabolisme.
- Pharmacocinétiques de population
Une analyse de pharmacocinétique de population de patients atteints de LMC a montré une légère influence de l'âge sur le volume de distribution (augmentation de 12 % chez les patients > 65 ans), mais cette variation ne semble pas cliniquement significative e Bien que l'effet du poids corporel sur la clairance de l'imatinib laisse attendre une clairance moyenne de 8,5 l/h pour un patient pesant 50 kg, contre 11,8 l/h pour un patient pesant 100 kg, une adaptation de la posologie en fonction du poids n'est pas requise. Le sexe n'a aucune influence sur les paramètres cinétiques de l'imatinib.
- Pharmacocinétique chez l'enfant
Comme chez l'adulte, l'imatinib a été rapidement absorbé après administration orale chez le patient pédiatrique dans des études de phase I et de phase II. Chez l'enfant, l'administration de doses de 260 et 340 mg/m2/jour a permis d'obtenir des concentrations plasmatiques équivalentes au/ doses de respectivement 400 mg et 600 mg chez I adulte. La comparaison de l'ASC0-24, à J8 et J1 pour une dose de 340 mg/m2/jour a révélé une accumulation de 1,7 fois après des prises uniques quotidiennes itératives.
• Altération des fonctions organiques
L'imatinib et ses métabolites ne sont pas excrétés de façon significative par le rein. Bien que l'analyse des résultats pharmacocinétiques ait montré une variabilité interindividuelle considérable, l'exposition moyenne à l'imatinib n'était pas augmentée chez des patients qui présentaient un6 altération de la fonction hépatique à des degrés variables par comparaison au/ patients ayant une fonction hépatique normale (cf. Posologie et mode d'administration, Mises en garde spéciales et Précautions particulières d'emploi et Effets indésirables).
DONNÉES DE SÉCURITÉ PRÉCLINIQUE
Le profil de tolérance préclinique de l'imatinib a été évalué chez le rat, le chien, le singe et le lapin. Des études de toxicité à doses multiples ont mis en évidence des modifications hématologiques légères a modérées chez le rat, le chien et le singe avec des modifications de la moelle osseuse chez le rat et le chien. Le foie est un organe cible chez le rat et le chien. Des augmentations faibles à modérées des transaminases et de légères diminutions des taux de cholestérol, triglycérides, protéines totales et albumine ont été observées chez les deux espèces. Aucune modification histopathologique n'a été mise en évidence sur le foie de rat. Une toxicité hépatique sévère a été observée chez des chiens traités pendant deux semaines, avec une élévation des enzymes hépatiques, une nécrose hépato-cellulaire, une nécrose des canaux biliaires et une hyperplasie des canaux biliaires. Une toxicité rénale a été observée chez des singes traités pendant deux semaines, avec une minéralisation et une dilatation focales des tubules rénaux et une néphrose tubulaire. Une augmentation de l'urée sanguine (BUN) et de la créatinine a été observée chez plusieurs de ces animaux. Chez les rats une hyperplasie de l'épithélium de transition dans la papille rénale et dans la vessie a été observée à des doses > 6 mg/kg dans l'étude de 13 semaines, sans modification des paramètres urinaires et sanguins. Une augmentation du nombre d'infections opportunistes a été observée avec le traitement chronique par l'imatinib Dans une étude de 39 semaines chez le singe, la dose dépourvue d'effet indésirable observable n'a pu être définie avec la plus faible dose de 15 mg/kg, correspondant approximativement à un tiers de la dose maximale de 800 mg chez I homme basée sur la surface corporelle. Le traitement a entraîné une aggravation des infections paludéennes normalement réprimées chez ces animaux. L'imatinib n'a pas été considéré comme génotoxique dans un test sur cellules bactériennes in vitro (test d'AMES) dans un test sur cellules de mammifères in vitro (lymphome de souris) et dans un test sur micronoyaux de rat in vivo. Toutefois, des effets génotoxiques positifs ont été obtenus avec l'imatinib dans un test de clastogenèse (aberration chromosomique) sur cellules de mammifères in vitro (cellules ovariennes de hamster chinois) avec activation métabolique.
Deux intermédiaires de synthèse, présents dans le produit final, sont positifs au test de mutagenese d'AMES. Lun de ces intermédiaires était aussi positif dans le test sur le lymphome de souris. Dans une étude de fertilité, chez le rat mâle traité pendant 70 j avant accouplement, le poids des testicules et de l'épididyme et le pourcentage de mobilité des spermatozoïdes ont diminué à la dose de 60 mg/kg, approximativement équivalente à la dose clinique maximale de 800 mg/j, basée sur la surface corporelle. Cela n'a pas été observé à des doses 120 mg/kg. Une réduction légère à modérée de la spermatogenèse a aussi été observée chez le chien à des doses orales > 30 mg/kg. Chez des rats femelles traitées pendant 14 jours avant accouplement et pendant 6 jours de gestation, aucun effet n'a été observé sur l'accouplement ou sur le nombre de femelles gestantes. Par contre, à la dose de 60 mg/kg, les rats femelles ont présenté une perte foetale post-implantation significative et un nombre de foetus vivants réduit significativement. Ceci n'a pas été observé à des doses s 20 mg/kg.
Après administration orale au cours d'une étude sur le développement prénatal et post-natal chez le rat un écoulement vaginal rouge a été observé dans le groupe sous 45 mg/kg/jour au 14-15eme jour de gestation. A la même dose, le nombre de ratons mort-nés ou décédant au cours des 4 premiers jours du post-partum était plus élevé. Dans la descendance F„ à la même dose, les poids moyens étaient réduits de la naissance jusqu'au sacrifice final et le nombre de portées atteignant le critère de séparation prépuciale était légèrement plus faible La fertilité de la descendance F. n'était pas modifiée alors qu'un nombre accru de résorptions foetales et une diminution du nombre de foetus viables étaient observés à 45 mg/kg/jour. La dose non toxique pour les mères et la génération F, était de 15 mg/kg/ jour (soit un quart de la dose maximale humaine de 800 mg). L'imatinib est tératogène chez les rats lorsqu'il est administré au cours de l'organogenèse, à des doses >100 mg/kg, approximativement équivalente à la dose clinique maximale de 800 mg/jour, basée sar la surface corporelle Les effets tératogènes observés sont' une exencéphalie, une encéphalocèle, une réduction/absence de l'os frontal et une absence des os pariétaux. Ces effets n'ont pas été observés à des doses ≤ 30 mg/kg. Dans une étude de carcinogénicité d'une durée de deux ans menée chez le rat avec imatinib administré à la dose de 15, 30 et 60 mg/kg/j, une réduction statistiquement significative de la longévité a été observée chez les mâles à la dose de 60 mg/kg/j et chez les femelles à une dose ≥ 30 mg/kg/j. L'e/amen histo-pathologique des animaux a mis en évidence comme cause principale de décès ou de sacrifice des cardiomyopathies (pour les deux sexes), des néphropathies chroniques en progression (chez les femmes) et des papillomes des glandes prepuciales Les organes cibles des modifications néoplasiques étaient les reins, la vessie, l'urètre, les glandes prépuciales et clitoridiennes, l'intestin grêle, les glandes parathyroides, les glandes surrénales, et l'estomac (hors tissu glandulaire) Des papillomes/carcinomes des glandes prépuciales et clitoridiennes ont été observés à partir des doses de 30 mg/kg/j représentant approximativement 0,5 ou 0,3 fois l'e/position journalière (basée sur l'ASC) chez l'homme traite par 400 mg/j ou 800 mg/j respectivement et 0,4 fois l'exposition journalière (basée sur l'ASC) chez l'enfant traité par 340 mg/m2/jour. La dose sans effet observable (DSEO) était de 15 mg/kg/j. Les adénomes/carcinomes rénaux et les papillomes de la vessie et de l'urètre les adénocarcinomes de l'intestin grêle, les adénomes des parathyroïdes, les tumeurs médullaires bénignes et malignes des glandes surrénales et les carcinomes/papillomes de l'estomac (hors tissu glandulaire) ont été observés a la dose de 60 mg/kg/j' représentant approximativement 1,7 ou 1 fois l'e/position journalière (basée sur l'ASC) chez l'homme traité par 400 mg/j ou 800 mg/j respectivement et 1,2 fois l'e/position journalière (basée sur l'ASC) chez l'enfant traite par 340 mg/m2/jour. La dose sans effet observable (DSEO) était de 30 mg/kg/j. Le mécanisme et la pertinence chez l'homme des résultats de l'étude de carcinogénicité menée chez le rat ne sont pas encore clarifiés. Des lésions non-néoplasiques qui n'avaient pas été identifiées au cours d'études précliniques antérieures, ont été observées sur le système cardiovasculaire, le pancréas, les glandes endocrines et les dents. Les modifications les plus importantes comprenaient l'hypertrophie et la dilatation cardiaque responsables de signes d'insuffisance cardiaque.