INVANZ
INVANZ®
Ertapénème.
IDENTIFICATION DU MEDICAMENT
FORMES ET PRÉSENTATIONS
Poudre pour solution à diluer pour perfusion.
Poudre blanche à blanchâtre.
COMPOSITION
Chaque flacon contient 1,0g d'ertapénème équivalent à 1,046 g d'ertapénème sodique, équivalent à 1,0 g d'ertapénème.
Excipients : Chaque dose de 1,0 g contient environ 6,0 mEq de sodium (environ 137 mg). Bicarbonate de sodium (E500). hydroxyde de sodium (E524) pour ajuster le ph à 7,5.
CLASSE PHAMACOTHERAPEUTIQUE
Carbapénèmes. code ATC : 01D 03
INDICATIONS
Traitement des infections suivantes lorsqu'elles sont dues à des espèces bactériennes connues pour être sensibles ou possiblement sensibles à l'ertapénème et lorsqu'un traitement parentéral est nécessaire (cf. "Mises en gardes et precautions d'emploi" et "Pharmacodynamie")
• Infections intra-abdominales
• Pneumonies communautaires
• Infections gynécologiques aiguës
• Infections de la peau et des tissus mous du pied chez le diabétique (voir rubrique 4.4).
POSOLOGIE ET MODE D'ADMINISTRATION
POSOLOGIE
• Adultes et adolescents (13 à 17 ans) : la dose d'INVANZ est de1 gramme (g), administré une fois par jour par voie intraveineuse (cf. "Précautions particulières de manipulation et d'élimination").
• Nourrissons et Enfants (3 mois à 12 ans): la dose d'INVANZ est de 15mg/kg, administrés 2 fois/jour (ne pas dépasser 1 g/jour) par voie intraveineuse (cf. "Précautions particulières de manipulation et d'élimination").
INVANZ est déconseillé chez l'enfant de moins de 3 mois en l'absence de donnée de sécurité d'emploi et d'efficacité (cf. "Mises engarde spéciales et précautions d'emploi"et "Pharmacocinétiques").
• Insuffisance rénale : INVANZ peut être utilisé chez des patients adultes ayant une insuffisance rénale.
Chez les patients dont la clairance de la créatinine est > 30ml/min/1,73m2, aucune adaptation posologique n'est nécessaire. Chez des patients ayant une insuffisance rénale sévère les données de tolérance et d'efficacité de l'ertapénème sont insuffisantes pour justifier une recommandation posologique.
Par conséquent l'ertapénème ne doit pas être utilisé chez ces patients (cf. "Pharmacocinétiques").
Aucune donnée n'est disponible chez l'enfant et l'adolescent ayant une insuffisance rénale.
• Patients sous hémodialyse : Chez les patients hémodialysés.les données de tolérance et d'efficacité de l'ertapénème sont insuffisantes pour justifier une recommandation posologique. Par conséquent, l'ertapénèmene ne doit pas être utilisé chez ces patients.
• Insuffisance hépatique : Aucune adaptation posologique n'est recommandée chez les patients insuffisants hépatiques (cf. "Pharmacocinétiques").
• Patients âgés: La dose recommandée d'INVANZ sera administrée, sauf en cas d'insuffisance rénale sévère (voir Insuffisance rénale).
MODE D'ADMINISTRATION
Administration intraveineuse
INVANZ doit être perfusé pendant 30 minutes. La durée habituelle du traitement par INVANZ est de 3 à14 jours, mais elle peut varier selon le type et la sévérité de l'infection et du (des) pathogène(s) en cause. Il est possible de poursuivre le traitement par un antibiotique oral approprié s'il état clinique le permet.
Précautions particulières de manipulation et d'élimination.
Pour usage unique seulement.
Les solutions reconstituées seront diluées dans une solution de chlorure de sodium à 9 mg/ml (0,9%) immédiatement après préparation.
Préparation pour administration intraveineuse: INVANZ doit être reconstitué puis dilué avant administration.
• Adultes et adolescents (13 à 17 ans) :
1. Reconstitution:
Reconstituer le contenu d'un flacon de 1g d'INVANZ avec 10 ml d'eau pour préparations injectables ou avec une solution de chlorure de sodium à 9mg/ml (0,9%) pour obtenir une solution reconstituée de 100mg/ml approximativement. Bien agiter pour dissoudre (voir rubrique 6.4).
2. Dilution:
''- Pour une poche de diluant de 50 ml: Pour une dose d'1 g, transférer immédiatement le contenu du flacon reconstitué dans une poche de 50 ml d'une solution de chlorure de sodium à 9 mg/ml (0,9%); ou
- Pour un flacon de diluant de 50 ml: Pour une dose d'1g, retirer 10 ml d'un flacon de 50 ml d'une solution de chlorure de sodium à 9 mg/ml (0,9%) et les jeter. Transférer le contenu du flacon de 1g d'INVANZ reconstitué dans le flacon de 50ml de la solution de chlorure de sodium à 9mg/ml (0,9%).
3. Perfusion :
Perfuser sur une période de 30 minutes.
• Enfants (3 mois à 12 ans):
1. Reconstitution:
Reconstituer le contenu d'un flacon de 1g d'INVANZ avec 10 ml d'eau pour préparations injectables ou avec une solution de chlorure de sodium à 9 mg/ml (0,9%) pour obtenir une solution reconstituée de 100mg/ml approximativement. Bien agiter pour dissoudre (voir rubrique 6.4).
2. Dilution :
- Pour une poche de diluant: transférer un volume équivalent à 15mg/kg de poids corporel (ne pas dépasser 1 g/jour) dans une poche d'une solution de chlorure de sodium à 9mg/ml (0,9%) pour une concentration finale de 20mg/ml ou moins; ou
- Pour un flacon de diluant : transférer un volume équivalent à 15mg/kg de poids corporel (ne pas dépasser 1 g/jour) dans un flacon de solution de chlorure de sodium à 9mg/ml (0,9%) pour une concentration finale de 20mg/ml ou moins.
3. Perfusion :
Perfuser sur une période de 30 minutes. La compatibilité d'INVANZ avec des solutions intraveineuses contenant de l'héparine sodique et du chlorure de potassium a été démontrée. Les solutions reconstituées seront examinées visuellement afin de rechercher la présence de particules ou d'une coloration anormale avant l'administration, si l'emballage extérieur le permet. Les solutions d'INVANZ sont incolores à jaune pâle. Les variations de couleur au sein de cette gamme n'affectent pas l'activité. Tout produit non utilisé ou déchet doit être éliminé conformément à la réglementation en vigueur.
CONTRE-INDICATIONS
- Hypersensibilité à la substance active ou à l'un des excipients
- Hypersensibilité à tout autre antibactérien du groupe des carbapénèmes (par exemple, réactions anaphylactiques, réaction cutanée sévère) à tout autre antibiotique de la famille des bêtalactamines (par exemple, pénicillines ou céphalosporines).
MISES EN GARDE et PRÉCAUTIONS D'EMPLOI
Des réactions d'hypersensibilité (anaphylactiques) graves et parfois fatales ont été rapportées chez des patients traités par bêtalactamines. La survenue de ces réactions est plus probable chez de patients ayant des antécédents d'hypersensibilités à de multiples allergènes. Avant de débuter un traitement par l'ertapénème, l'interrogatoire doit rechercher notamment des antécédents de réactions d'hypersensibilités pénicillines, céphalosporines, autres bêtalactamines ainsi qu'autres allergènes (cf. "Contre-indications"). La survenue d'une réaction allergique à l'ertapénème impose l'arrêt immédiat du traitement.
Des réactions anaphylactiques graves nécessitent l'instauration immédiate d'un traitement d'urgence.
Comme pour les autres antibiotiques, l'utilisation prolongée d'ertapénème peut entrainer la prolifération de germes résistants. La réévaluation de l'état du patient est essentielle. En cas de survenue d'une surinfection au cours du traitement, des mesures appropriées seront prises. Des colites « associées aux antibiotiques » et des colites pseudomembraneuses ont été rapportées avec presque tous les antibiotiques, y compris l'ertapénème, dont la sévérité peut varier d'une forme légère jusqu'à celle mettant en jeu le pronostic vital. Cediagnostic doit être envisagé chez les patients présentant une diarrhée secondaire à l'administration d'antibiotiques. Un arrêt du traitement avec INVANZ et l'administration d'un traitement spécifique contre Clostridium difficile seront envisagés.
Des médicaments inhibant le péristaltisme ne doivent pas être administrés.
Des convulsions ont été rapportés lors des études cliniques chez des patients adultes au cours du traitement par l'ertapénème sodique (1g par jour) ou dans les 14 jours suivant l'arrêt du traitement. Les convulsions sont survenues plus fréquemment chez les patients âgés et chez les patients présentant des troubles préexistants du système nerveux central (par exemple: lésions cérébrales ou antécédent de convulsions) et/ ou une fonction rénale altérée. Des observations similaires ont été faites depuis la commercialisation.
L'efficacité d'INVANZ dans le traitement des pneumonies communautaires dues à Streptoccus pneumoniae résistant à la pénicilline n'a pas été établie.
L'expérience concernant l'utilisation de l'ertapénème chez les enfants de moins de 2 ans est limitée. Il faudra porter une attention particulière dans cette tranche d'âge en analysant la sensibilité du (des germes) en cause à l'ertapénème. Aucune donnée n'est disponible chez l'enfant de moins de 3 mois.
L'expérience concernant l'utilisation de l'ertapénème dans le traitement des infections sévères est limitée. Lors
des essais cliniques menés dans le traitement des pneumonies communautaires, chez l'adulte, 25% des patients évaluables traités par ertapénème avaient une maladie sévère (définie par un index de sévérité de la pneumonie >lll). Lors d'une étude clinique menée dans le traitement des infections gynécologiques aigu s, chez l'adulte, 26 % patients évaluables traités par ertapénème avaient une maladie sévère (définie par une température 39°C et/ou une bactériémie) 10 patients avaient une septicémie. Lors d'une étude clinique menée dans le traitement des infections intra-abdominales, chez l'adulte, 30% des patients évaluables traités par ertapénème avaient une péritonite généralisée et 39% présentaient des infections de sites autres que l'appendice, incluant l'estomac, le duodénum, l'intestin grêle, le côlon, et la vésicule biliaire; le nombre de patients évaluables inclus dans des scores APAC E ll>15 était limité et l'efficacité chez ces patients n'a pas été établie. L'efficacité de l'ertapénème dans le traitement des infections du pied chez le diabétique en cas d'ostéomyélite concomitante n'a pas été établie.
CONDUITE ET UTILISATION DE MACHINES
Aucune étude relative aux effets sur l'aptitude à conduire des véhicules et à utiliser des machines n'a été menée. INVANZ peut influencer la capacité des patients à conduire des véhicules ou à utiliser des machines. Les patients doivent être informés que des cas d'étourdissements et de somnolence ont été rapportés avec INVANZ (cf. Effets indésirables).
INTERACTIONS
Il n'est pas attendu d'interaction résultant d'une inhibition de la clairance de médicaments, que cette clairance
soit médiée par la glycoprotéine-P ou par les cytochromes (cf. "Pharmacocinétique" ).
Les antibiotiques pénèmes et carbapénèmes peuvent diminuer les taux plasmatiques de l'acide valproïque. Un contrôle des taux plasmatiques de l'acide valproïque doit être envisagé si l'ertapénème est administré avec de l'acide valproïque.
Incompatibilités:
• Ne pas utiliser de solvants ou de liquides pour perfusion contenant du dextrose pour reconstituer ou administrer l'ertapénème sodique.
• En l'absence d'études de compatibilité, ce médicament ne doit pas être mélangé à d'autres médicaments. 6.3 Durée de conservation.2 ans.
• Après reconstitution:
- Les solutions diluées seront utilisées immédiatement. Si elles ne sont pas utilisées immédiatement, l'utilisateur en sera responsable.
- Les solutions diluées (approximativement 20mg/ml d'ertapénème) sont physiquement et chimiquement stables pendant 6 heures à température ambiante (25°C) ou pendant 24 heures de 2 à 8°C (au réfrigérateur).
- Les solutions seront utilisées dans les 4 heures après avoir été sorties du réfrigérateur.
GROSSESSE et ALLAITEMENT
Des études correctes et contrôlées n'ont pas été réalisées au cours de la grossesse. Les études réalisées chez l'animal n'ont pas mis en évidence d'effet directement ou indirectement nocif sur le déroulement de la grossesse, le développement embryo-foetal, la mise bas ou le développement post-natal. Cependant l'ertapénème ne doit pas être prescrit au cours de la grossesse, sauf si le bénéfice maternel est supérieur au risque potentiel pour le foetus. L'ertapénème est excrété dans le lait maternel. En raison de la survenue possible de réactions indésirables chez l'enfant allaité,les mères traitées par INVANZ ne doivent pas allaiter.
EFFETS INDÉSIRABLES
• Adultes de 18ans et plus:
Au total, plus de 2200 patients ont été traités par l'ertapénème lors des études cliniques, dont plus de 2150 à la dose de 1g d'ertapénème. Des effets indésirables (c'est à dire considérés par l'investigateur comme possiblement, probablement, ou assurément reliés à la prise du médicament) ont été rapportés chez environ 20% des patients traités par l'ertapénème. Le traitement a été arrêté en raison de la survenue d'effets indésirables chez 1,3% des patients. Dans une étude clinique additionnelle, 476 patients ont reçu, dans le cadre d'une prophylaxie des infections postopératoires en chirurgie
colorectale, 1 g d'ertapénème en dose unique avant l'incision chirurgicale.
Pour les patients n'ayant reçu qu'INVANZ, les effets indésirables les plus fréquemment rapportés au cours du traitement et jusqu'à 14 jours après l'arrêt ont été: diarrhée (4,8%), complication au niveau de la veine perfusée
(4,5%),et nausées (2,8%).
Pour les patients n'ayant reçu qu'INVANZ, les anomalies biologiques les plus fréquemment rapportées au cours du traitement et jusqu'à 14 jours après l'arrêt ont été: élévation du taux d'ALAT (4,6%),du taux d'ASAT (4,6%),des phosphatases alcalines (3,8%)et des plaquettes (3,0%).
• Enfants et adolescents (de 3mois à 17 ans) :
Au total 384 patients ont été traités par l'ertapénème lors des études cliniques. Le profil de sécurité d'emploi est comparable à celui des adultes. Des effets indésirables (c'est-à-dire jugés par les investigateurs comme possiblement, probablement ou assurément reliés à la prise du médicament) ont été rapportés chez environ 20,8% des patients traités par l'ertapénème. Le traitement a été interrompu en raison d'effets indésirables chez 0,5% des patients.
Pour les patients n'ayant reçu qu'INVANZ, les effets indésirables les plus fréquemment rapportés au cours du traitement et jusqu'à 14 jours après son arrêt ont été: diarrhée (5,2%) et douleur au point de perfusion (6,1%).
Pour les patients n'ayant reçu qu'INVANZ, les anomalies biologiques les plus fréquemment rapportées au cours du traitement et jusqu'à 14 jours après l'arrêt ont été: diminution du taux de polynucléaires neutrophiles (3,0%) et augmentation du taux d'ALAT (2,9%) et d'ASAT (2,8%).
Pour les patients n'ayant reçu qu'INVANZ, les effets indésirables suivants rapportés au cours du traitement et jusqu'au jours après l'arrêt ont été 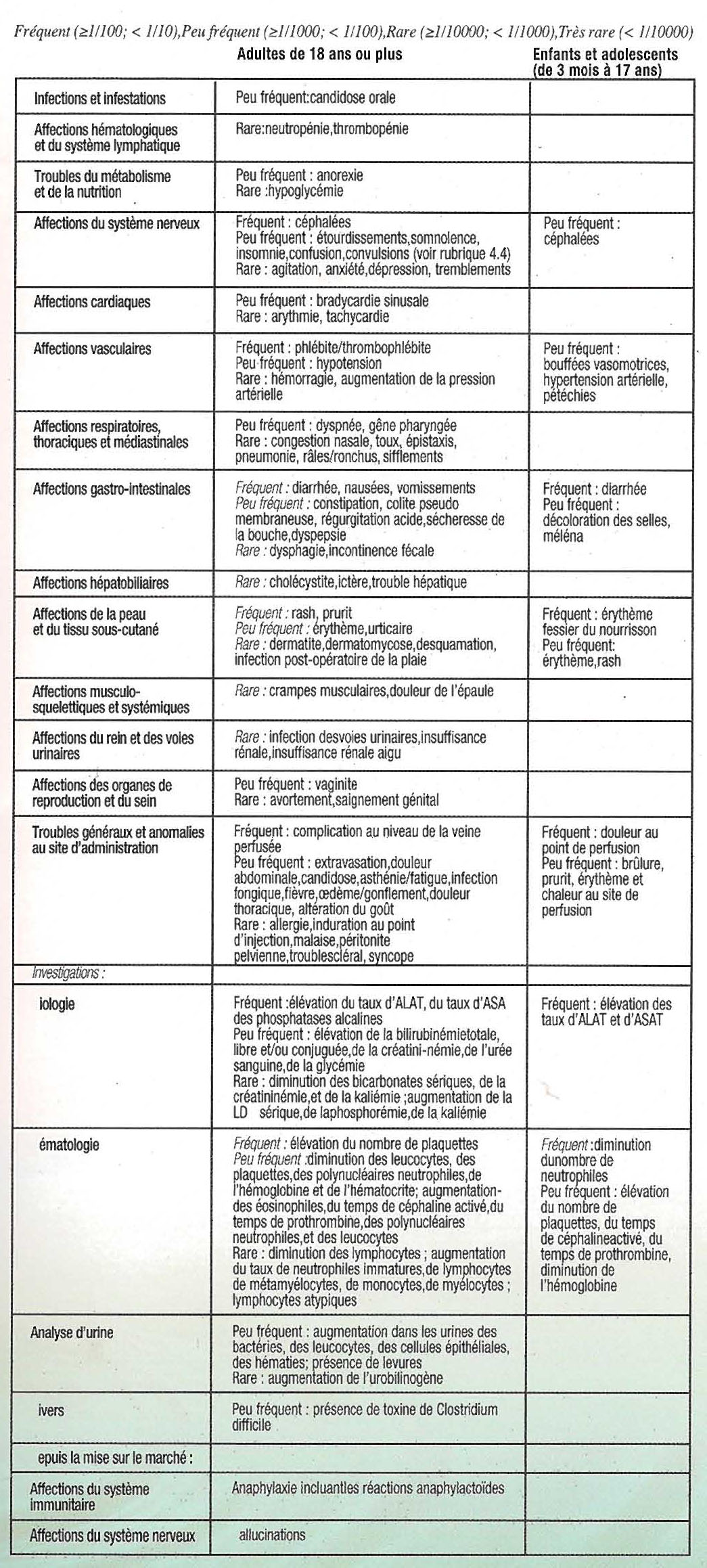
SURDOSAGE
Aucune donnée spécifique n'est disponible sur le traitement d'un surdosage par l'ertapénème. Le surdosage en ertapénème est peu probable.
L'administration intraveineuse d'ertapénème à une dose quotidienne de 3g pendant 8 jours à des volontaires sains adultes n'a pas entraîné de toxicité significative. Au cours des études cliniques chez l'adulte, l'administration accidentelle allant jusqu'à 3g par jour n'a pas provoqué d'effets indésirables
cliniquement importants. Dans les études cliniques pédiatriques, l'administration intraveineuse d'une dose unique de 40 mg/kg jusqu'à un maximum de 2 g, n'a pas entraîné de toxicité. Cependant, en cas de surdosage le traitement par INVANZ sera arrêté et un traitement symptomatique administré jusqu'à l'élimination rénale du médicament. L'ertapénème peut être éliminé partiellement par hémodialyse (cf. "Pharmacocinétiques"); toute fois,aucune information n'est disponible sur l'utilisation de l'hémodialyse pour traiter le surdosage.
PHARMACODYNAMIE
- Propriétés générales
• Mode d'action:
L'ertapénème inhibe la synthèse des parois cellulaires des bactéries après fixation aux protéines de liaison aux pénicillines (PLP). Pour Escherichia coli, l'affinité est la plus forte pour les PLP 2 et 3.
• Rapport pharmacocinétique /pharmacodynamie (PC/PD): Lors d'études précliniques de pharmacocinétique / pharmacodynamie, il a été démontré comme pour d'autres bêtalactamines, que le temps où la concentration plasmatique d'ertapénème excède la CMI pour la bactérie, était le meilleur paramètre prédictif de l'efficacité. Mécanisme de résistance. Pour les espèces considérées comme sensibles à l'ertapénème, les cas de résistance ont été peu fréquents dans les études de surveillance menées en Europe. Pour les isolats résistants, une résistance à d'autres antibiotiques de la classe des carbapénèmes a été observée pour certaines souches mais pas pour toutes. L'ertapénème reste stable à l'hydrolyse par la plupart des bêta-lactamases y compris les pénicillinases, les céphalosporinases et les bêta-lactamases à spectre élargi, mais pas à l'hydrolyse par les métallo-bêta-lactamases. Les staphylocoques méticilline-résistants et les entérocoques sont résistants à l'ertapénème du fait de la non-sensibilité de la cible, les PLP; P.aeruginosa et d'autres bactéries non fermentantes sont généralement résistantes, probablement du fait d'une pénétration limitée et d'un efflux actif. La résistance n'est pas fréquente chez les entérobactéries et le médicament est généralement actif contre les entérobactéries produisant des bêta-lactamases à spectre élargi (BLSE). La résistance peut cependant être observée quand aux BLSE ou à d'autres bêta-lactamases très actives (par exemple de types AmpC) est associée une perméabilité réduite, due à la perte d'une ou plusieurs porines de la membrane externe, ou une régulation à la hausse de l'efflux. La résistance peut aussi survenir par l'acquisition de bêta-lactamases possédant une activité significative d'hydrolyse des carbapénèmes (par exemple les métallo-bêta-lactamases de type IMP, VIM ou KPC), mais celles-cisontrares. Le mécanisme d'action de l'ertapénème diffère de celui d'autres classes d'antibiotiques, comme les quinolones, les aminosides, les macrolides et les tétracyclines. Il n'existe pas de résistance croisée au niveau de la cible entre l'ertapénème et ces produits. Cependant, des micro-organismes peuvent présenter une résistance à plus d'une classe d'agents antibactériens quand l'imperméabilité à certains produits et/ou l'efflux actif, sont présents comme mécanisme de résistance.
- Concentrations critiques.
Les concentrations critiques de l'EUCAST sont les suivantes :
+ Entérobaéctries : S< 0,5 mg/l et R>1 mg
+ Streptococcus A,B,C,G: S<0,5mg/I et R>0,5mg/I
+ Streptococcus pneumoniae : S<0,5mg/I et R>0,5 mg/l» Haemophilus influenzae: S<0,5mg/let R>0,5 mg/l
+ M.catarrhalis: S<0,5mg/I et R>0,5mg/I» Anaérobies à Gram négatif : S < lmg/l et R> 1 mg
- Concentrations critiques non liées à l'espèce:
S < 0,5 mg/l et R>1 mg (NB : La sensibilité des staphylocoques à l'ertapénème est déduite de la sensibilité à la méticilline). Les prescripteurs sont informés que les concentrations critiques locales, si elles sont disponibles, doivent être consultées.
• Sensibilité microbiologique:
La prévalence de la résistance acquise peut varier en fonction de la région géographique et du temps pour certaines espèces. Il est donc utile de disposer d'informations sur la prévalence de résistance locale, surtout pour le traitement d'infections sévères. Des foyers localisés d'infections dues à des espèces résistantes aux carbapénèmes ont été rapportés dans l'Union Européenne. Les données ci-après fournissent une orientation sur les probabilités de la sensibilité d'une souche à l'ertapénème.

- Informations recueillies lors des études cliniques.
• Efficacité dans les études pédiatriques:
L'ertapénème a été évalué chez des patients âgés de 3 mois à 17 ans dans des études multicentriques, randomisées, comparatives, dont l'objectif principal était d'évaluer la tolérance de l'ertapénème, l'efficacité n'étant qu'un critère secondaire d'évaluation. Le tableau ci-contre donne la proportion de patients ayant une réponse clinique favorable lors de la visite de suivi après traitement, dans la population clinique en Intention deTraiter (ITT):
| Pathologies+ |
Tranche d'age |
Ertapénème
|
Ertapénème |
||
|
|
|
n/m |
% |
n/m |
% |
| Pneumonie Communautaire (PC)
|
3 à 23 mois
|
31/35
|
88,6
|
13/13
|
100,0 |
|
|
2 à 12 mois
|
55/57
|
96,5
|
16/17
|
94,1 |
|
|
13 à 17 ans
|
3/3
|
100.0
|
3/3
|
100,0 |
| Pathologies
|
Tranche d'age
|
Ertapénème
|
Ertapénème |
||
|
|
|
n/m
|
%
|
n/m
|
% |
| Infections intra-abdominales (HA)
|
2 à 12 ans
|
28/34
|
82,4
|
7/9
|
77,8 |
|
|
13 à 17 ans
|
15/16
|
93,8
|
4/6
|
66,7 |
| Infections pelviennes aigues (IAP)
|
13 à 17 ans
|
25/25
|
100,0
|
8/8
|
100,0 |
+ : Inclusion de 9 patients dans le groupe ertapénème (7 PC et 2 HA), 2 patients dans le groupe ceftriaxone (2 PC), et 1 patient avec HA dans le groupe ticarcilline/acide clavulanique, présentant une bactériémie après l'entrée dans l'étude
PHARMACOCINETIQUE
• Concentrations plasmatiques:
Les concentrations plasmatiques moyennes d'ertapénème après une perfusion intraveineuse unique de 30 minutes d'une dose de 1 g chez des adultes jeunes sains (âgés de 25 à 45 ans) étaient de 155 microgrammes/ml (Cmax) à 0,5 heure après traitement (fin de la perfusion), 9 microgrammes/ml 12 heures après traitements 1 microgramme/ml 24 heures après traitement. L'aire sous la courbe de la concentration plasmatique (ASC) d'ertapénème chez l'adulte augmente presque proportionnellement à la dose sur l'intervalle de doses 0,5 à 2g. Il n'y a pas d'accumulation d'ertapénème chez l'adulte après l'administration de doses intraveineuses multiples allant de 0,5 à 2 g par jour. Les concentrations plasmatiques moyennes d'ertapénème après une perfusion intraveineuse unique de 30 minutes d'une dose de 15mg/kg (jusqu'à une dose maximum de 1 g) chez des patients âgés de 3 à 23 mois étaient de 103,8 microgrammes/ml (Cmax) 0,5 heure après traitement (fin de la perfusion), 13,5 microgrammes/ml 6 heures après traitement, et 2,5 microgrammes/ml 12 heures après traitement. Les concentrations plasmatiques moyennes d'ertapénème après une perfusion intraveineuse unique de 30 minutes d'une dose de 15mg/kg (jusqu'à une dose maximum de 1g) chez des patients âgés de 2 à 12 ans étaient de 113,2 microgrammes/ml (Cmax) 0,5 heure après traitement (fin de la perfusion), 12,8 microgrammes/ml 6 heures après traitement, et 3,0 microgrammes/ml 12 heures après traitement. Les concentrations plasmatiques moyennes d'ertapénème après une perfusion intraveineuse unique de 30 minutes d'une dose de 20mg/kg (jusqu'à une dose maximum de 1g) chez des patients âgés de 13 à 17 ans étaient de 170,4 microgrammes/ml (Cmax) 0,5 heure après traitement (fin de la perfusion), 7,0 microgrammes/ml 12 heures après traitement et 1,1 microgramme/ml 24 heures après traitement. Les concentrations plasmatiques moyennes d'ertapénème après une perfusion intraveineuse unique de 30 minutes d'une dose de1g chez trois patients âgés de 13 à 17 ans étaient de 155,9 microgrammes/ml (Cmax) 0,5 heure après traitement (fin de la perfusion), et 6,2 microgrammes/ml 12 heures après traitement.
• Distribution:
Chez l'homme, l'ertapénème est fortement lié aux protéines plasmatiques. Chez des adultes jeunes sains (âgés de 25 à 45 ans), la liaison aux protéines de l'ertapénème diminue à mesure que la concentration plasmatique augmente, passant de 95% environ pour une concentration plasmatique approximatives microgrammes/ml à 92% environ pour une concentration plasmatique approximative de 155 microgrammes/ml (concentration moyenne obtenue à la fin d'une perfusion de 1 g par voie intraveineuse). Le volume de distribution (Vdss) de Pertapénème est d'environ 8 litres chez l'adulte (0,11 litre/kg); d'environ 0,2 litre/kg chez les enfants âgés de 3 mois à 12ans et d'environ 0,16 Iitre/kg chez les adolescents âgés de 13 à 17 ans les concentrations d'ertapénème obtenues chez l'adulte dans le liquide phlycténulaire à chaque temps de prélèvement le troisième jour après une administration intraveineuse quotidienne d'une dose de 1g ont montré un ratio de l'ASC dans le liquide phlycténulaire: l'ASC dans le plasma est de 0,61.Des études in-vitro indiquent que Pertapénème ne déplace pas d'une façon notable les médicaments fortement liés aux protéines circulantes (warfarine, éthinylestradiol. et noréthindrone). La modification de la liaison était <12% au pic de concentration plasmatique de l'ertapénème après une dose de 1g. ln-vivo, le probénécide (500 mg toutes les 6 heures) a diminué de 91% approximativement à 87% approximativement la fraction de liaison de l'ertapénème dans le plasma à la fin de la perfusion chez des sujets ayant reçu une dose unique intraveineuse de 1 g. Les effets de cette modification sont probablement transitoires. Une interaction cliniquement significative due au déplacement de l'ertapénème par un autre médicament ou au déplacement d'un autre médicament par l'ertapénème est peu probable. Les études in-vitro indiquent que l'ertapénème n'inhibe pas le transport de la digoxine ou de la vinblastine par la glycoprotéine P et que l'ertapénème n'est pas un substrat pour la glycoprotéine P.
• Métabolisme:
Chez des adultes jeunes sains (âgés de 23 à 49 ans), après perfusion intraveineuse de 1g d'ertapénème radiomarqué, la radioactivité plasmatique est essentiellement (94%) composée d'ertapénème. Le métabolite principal de l'ertapénème est le dérivé à cycle ouvert formé par hydrolyse du noyau bêtalactame par la déhydropeptidase-1. Les études in-vitro avec des microsomes hépatiques humains indiquent que l'ertapénème n'inhibe pas le métabolisme médié par les six principales isoformes du CYP: 1A2, 2C9, 2C19, 2D6, 2E1 et 3A4.
• Elimination:
Après l'administration d'une dose intraveineuse de 1g d'ertapénème radiomarqué à des adultes jeunes sains (âgés de 23 à 49 ans),environ 80% sont retrouvés dans les urines et 10% dans les fèces. Sur 80% retrouvés dans les urines, environ 38% sont éliminés sous forme inchangée d'ertapénème et environ 37% sous forme de métabolite à cycle ouvert Chez des adultes jeunes sains (âgés de 18 à 49 ans) et des patients âgés de 13 à 17 ans, traités par une dose intraveineuse de 1 g, la demi-vie plasmatique moyenne est d'environ 4 heures la demi-vie plasmatique moyenne chez les enfants âgés de 3 mois à 12 ans est d'environ 2,5 heures. Les concentrations moyennes d'ertapénème retrouvées dans les urines dépassent 984 microgrammes/ml au cours de la période 0 à 2 heures après l'administration et elles dépassent 52 microgrammes/ml au cours de la période 12 à 24 heures après l'administration.
• Populations spéciales.
- Sexe: Les concentrations plasmatiques d'ertapénème sont comparables chez l'homme et la femme
- Personnes âgées: Après l'administration d'une dose intraveineuse de 1g et de 2g d'ertapénème, les concentrations plasmatiques sont légèrement plus élevées (environ 39% et 22%, respectivement) chez les personnes âgées saines (>65 ans) que chez les adultes plus jeunes (<65 ans). En l'absence d'insuffisance rénale sévère, aucune adaptation posologique n'est nécessaire chez les patients âgés.
- Enfants et adolescents: Les concentrations plasmatiques d'ertapénème sont comparables chez les patients adolescents de 13 à 17 ans et les adultes après l'administration d'une dose intraveineuse de 1g une fois par jour. Les paramètres pharmacocinétiques étaient généralement comparables chez des patients âgés de 13 à17 ans et chez des jeunes adultes sains, après administration d'une dose de 20mg/kg (jusqu'à une dose maximum de 1 g). Afin d'estimer les données pharmacocinétiques dans le cas où tous les patients de cette tranche d'âge avaient reçu une dose de1g, celles-ci ont été calculées en ajustant la dose à 1g, en supposant une linéarité. Une comparaison des résultats montre que le profil pharmacocinétique obtenu après une dose de 1 g une fois par jour chez des patients âgés de 13 à17 ans est comparable à celui des adultes. Les ratios (13 à 17 ans/adultes) pour l'ASC, la concentration en fin de perfusion et la concentration au milieu de l'intervalle de doses étaient de 0,99; 1,20 et 0,84 respectivement. Les concentrations plasmatiques au milieu de l'intervalle de doses après administration intraveineuse d'une dose de 15mg/kg d'ertapénème chez des patients âgés de 3 mois à 12 ans sont comparables aux concentrations plasmatiques au milieu de l'intervalle de doses après administration intraveineuse d'une dose de 1g une fois par jour chez des adultes (voir concentrations plasmatiques).La clairance plasmatique (ml/min/kg) de l'ertapénème chez des patients âgés de 3 mois à 12 ans est environ 2 fois plus élevée comparée à celle des adultes. A la dose de 15 mg/kg, la valeur de l'ASC et les concentrations plasmatiques au milieu de l'intervalle de dose chez des patients âgés de 3 à12 ans, étaient comparables à celles de jeunes adultes sains recevant une perfusion intraveineuse de 1g d'ertapénème.
- Insuffisance hépatique: La pharmacocinétique de l'ertapénème chez les patients ayant une insuffisance hépatique n'a pas été établie. En raison de l'ampleur limitée du métabolisme hépatique de l'ertapénème, aucune modification de la pharmacocinétique de l'ertapénème par insuffisance hépatique n'est attendue. Par conséquent, aucune adaptation posologique n'est recommandée chez les patients ayant une insuffisance hépatique.
- Insuffisance rénale: Après l'administration d'une dose unique intraveineuse de 1g d'ertapénème chez l'adulte, les ASC de l'ertapénème total (lié et non lié) et de Pertapénèmenon lié sont similaires chez les patients ayant une insuffisance rénale légère (Clcr:60 à 90ml/min/1,73m2) et chez les sujets sains (âgés de 25 à 82 ans).Les ASC de l'ertapénème total et de l'ertapénème non lié augmen¬tent chez les patients ayant une insuffisance rénale modérée (Clcr:31 à 59 ml/min/1,73m2) d'environ 1,5 fois et 1,8 fois, respectivement par rapport aux sujets sains. Les ASC de l'ertapénème total et de l'ertapénème non lié augmentent chez les patients ayant une insuffisance rénale sévère (Clcr: 5 à 30ml/min/1,73 m2) d'environ 2,6 fois et 3,4 fois, respectivement, par rapport aux sujets sains. Les ASC de l'ertapénème total et de l'ertapénème non lié augmentent chez les patients qui nécessitent une hémodialyse d'environ 2,9 fois et 6,0 fois, respectivement, entre les séances de dialyse, par rapport aux sujets sains. Après l'administration d'une dose unique intraveineuse de 1g immédiatement avant une séance d'hémodialyse, environ 30% de la dose administrée sont retrouvés dans le dialysat II n'y a pas de données chez les enfants et adolescents ayant une insuffisance rénale. Il n'y a pas de données appropriées de tolérance et d'efficacité de l'ertapénème chez des patients ayant une insuffisance rénale sévère et les patients nécessitant une hémodialyse pour justifier une recommandation posologique. Par conséquent, l'ertapénème ne doit pas être utilisé chez ces patients.
DONNÉES DE SÉCURITÉ PRÉCLINIQUES
Les données non cliniques issues des études conventionnelles de sécurité d'emploi de pharmacologie, de toxicité en administration réitérée, de génotoxicité, et des fonctions de reproduc¬tion, n'ont pas révélé de risque particulier pour l'homme. Cependant, une diminution du nombre despolynucléaires neutrophiles est survenue chez les rats ayant reçu des doses élevées d'ertapénème, cette diminution n'a pas été considérée comme étant un problème significatif de sécurité d'emploi. Aucune étude à long terme n'a été réalisée chez l'animal afin d'évaluer le potentiel carcinogène de Pertapénème.(E500). Hydroxyde de sodium (E524) pour ajuster le ph à 7,5.